获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往
原文由 lxdongzi2003 发表:
你的实验过程写的很详细,这样好分析。
如果你的样品在现有的流动相中不能全溶,可以试试降低乙腈的含量。(不太明白为什么要用乙腈,蛋白类物质应该可以用盐溶液直接溶解的。)
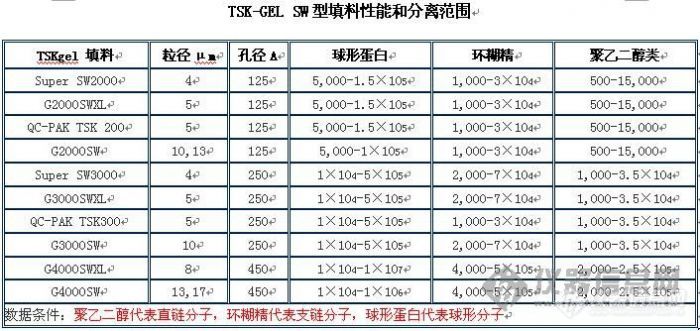
你的目标小肽的分子量在“主要集中在2000-300,少量在3000-5000”范围内,那你不应该选用G3000SWXL柱,该柱子的分离范围(球型分子)是1万到50万,大了,所以标准曲线有问题是正常的。
选用G2000SWXL能好些,它的分离范围是:5000到1.5万。
能分析分子量到几百的凝胶一般选用:SephadexG-15或G-10等。