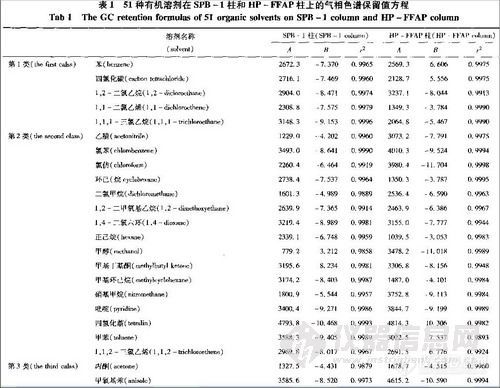
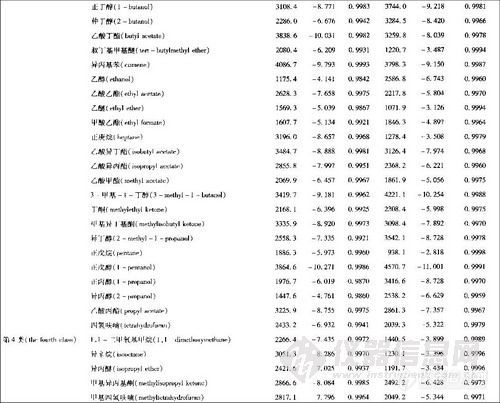
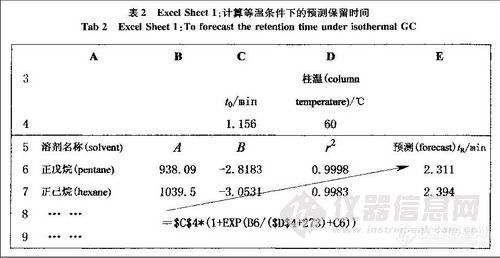
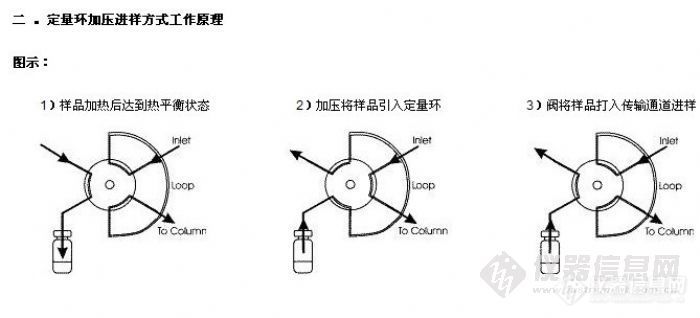
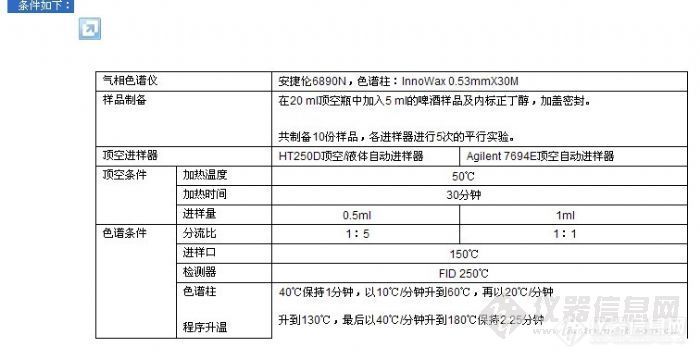
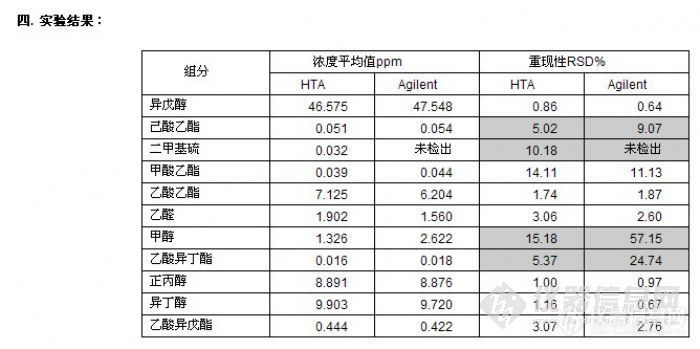
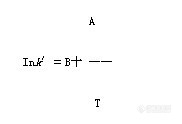顶空气相色谱系列讲座(127)(下)--啤酒中风味物质的顶空分析及评价 应用举例:
① GCO 法分析啤酒中的老化风味成分。
20ml 啤酒在隔氧避光条件下吸附在Extrelute 20 萃取柱上,再用100ml 戊烷解吸后蒸馏浓缩, 直接注入
气相色谱。
气相色谱条件: HP - 5 石英毛细管柱, 50 m 长, 0. 32mm I. D. ; 柱温40 ℃, 以3 ℃/ min 升温至270 ℃保持45min ; 柱出口洗脱分流比FID ∶气味检测器为10 ∶90 ;气味检测器工作时间60min 。GCO 法与GC - FID 结合可以分析啤酒中的高级醇、酯类、酚类及反- 2 - 壬烯醛、苯乙醛等老化风味醛类物质。
② 多维
气相色谱—气味检测法与GC - MS 技术结合测定啤酒、酿造过程及原料中的风味化合物和异味组分。
啤酒酿造过程中产生杀菌剂味,例如2 ,6 - 二氯苯酚,单靠GC - MS 技术,因分离度不好, 无法定性, 采用多维
气相色谱—气味检测技术可以解决这一难题。先将样品酒用乙醚为萃取剂采用固相萃取管萃取及浓缩, 然后利用HP - Innowax 毛细柱由GC 风味检测器在保留时间为63. 7min 进行杀菌剂味测定。再将分离出的样品吸附在Tenax TA 管上,采用Quadrex007 甲基硅酮毛细柱进行分离,由GC 气味检测器在保留时间为18. 3min 进行杀菌剂味测定, 最后由多维GC - MS 在相同分离条件下进行测定, 可鉴别出该种化合物是2 ,6 - 二氯苯酚。
2. 6 电子自旋共振( ESR) 技术改善啤酒的风味稳定性
利用电子自旋共振光谱技术,可以测定啤酒的内抗氧化活性及OH 基团产生活性。在生产实践中,该技术可对酿造工艺对啤酒风味稳定性的影响进行分析,达到预测和改善啤酒风味稳定性的目的。将ESR 技术应用于啤酒新鲜度管理中, 与化学发光技术、老化风味醛类测定技术相结合,可以定量分析啤酒的新鲜度,而且得到的新鲜度值与感官品评有很好的相关性。
3 啤酒中挥发性风味化合物的风味评价
啤酒风味特征评价方法的原理是,通过
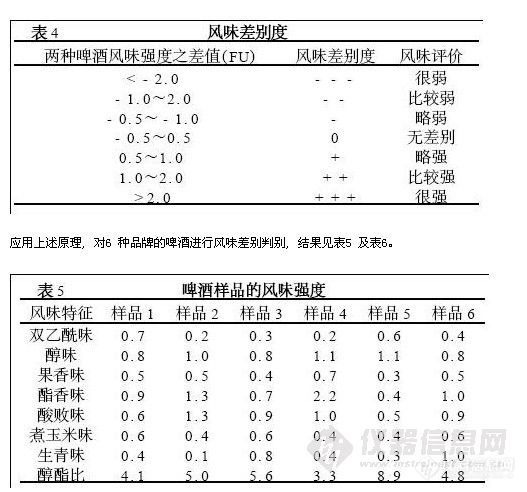
气相色谱对啤酒中的挥发性风味成分进行分析测定, 结合Meilgaad 有关风味阈值及风味协同作用的研究理论, 计算某种风味特征的风味强度值, 从而对啤酒的风味成分进行评价及对不同品牌啤酒进行风味差别的判别,达到控制和改善啤酒风味稳定性和一致性的目的。风味阈值是指某种风味成分在啤酒中可感受到的最低含量。某种风味成分对啤酒的影响,主要与其浓度和风味阈值有关,由此提出了风味强度的概念。风味强度( FU) = 风味物质浓度/ 风味阈值。根据风味强度值的大小, 将啤酒中的风味成分分为4类。
● 首要的风味成分( FU > 2. 0)
乙醇,酒花苦味物质(如异草酮) ,CO2。特殊啤酒:酒花香气成分,谷物香气成分,高浓啤酒的几种酯类和醇类,短链脂肪酸。
缺陷啤酒:反- 2 - 壬烯醛, 双乙酰和2 , 3 - 戊二酮, H2S、DMS 等含硫化合物, 乙酸, 3 - 甲基- 2 - 丁烯硫醇, 其他因微生物污染等生成的风味成分。
● 次要的风味成分(0. 5~2. 0 FU)
挥发性:香蕉酯(如乙酸异戊酯) ,苹果酯(如己酸乙酯) ,杂醇油(如异戊醇) ,C6、C8、C10脂肪酸,乙酸乙酯,丁酸和异戊酸,苯乙酸。
非挥发性:酚类,各种酸类,糖类及酒花化合物。
● 第三类风味成分(0. 1~0. 5 FU)
乙酸苯乙酯,对氨基苯乙酮,异戊醛,乙偶姻,γ- 戊内酯等。
● 其他风味成分( < 0. 1 FU)
指不在上述范围的风味成分。通过利用风味强度值这一概念, 可对啤酒风味成分进行评价。其有两方面的涵义:
3. 1 评价个体样品的风味特征,预测风味病害
一般来讲, 当某种风味物质的风味强度小于0. 5 时, 不会对风味产生影响;当风味强度在0. 5~2. 0 时,则会对啤酒风味产生一定的影响; 当风味强度大于2. 0 时, 对啤酒的风味有严重的影响。利用
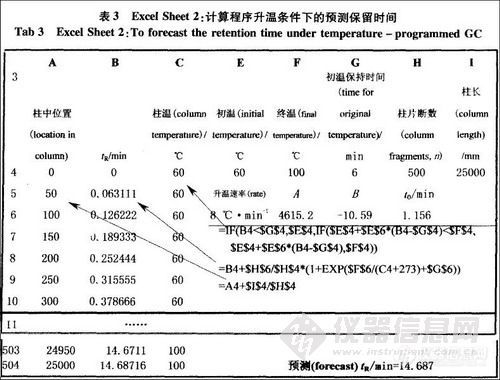
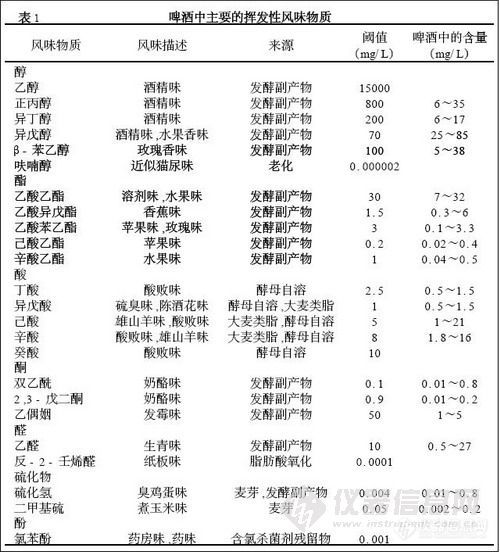
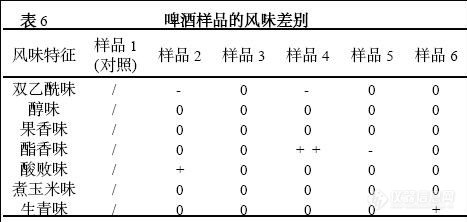
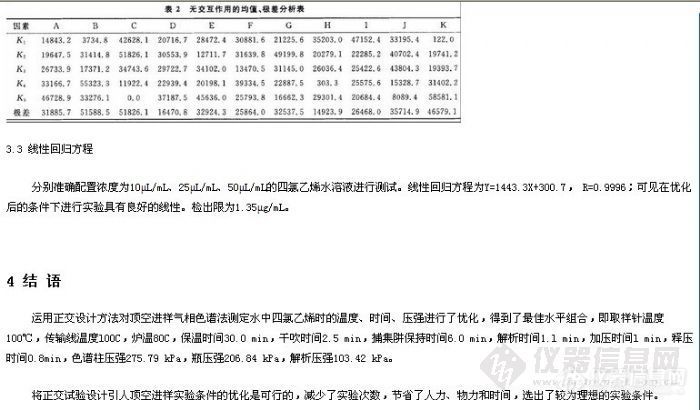
气相色谱分析方法,对啤酒中重要的高级醇、酯类、连二酮、乙醛、二甲基硫、脂肪酸等成分进行分析测定, 这些物质涵盖了啤酒的醇味、酯香味、双乙酰味、生青味、煮玉米味等风味特征,约占啤酒主要香气特征的50 % ,是啤酒香气成分的主要骨架。由于啤酒中的风味成分之间具有风味协同作用或风味累加作用, 在进行风味特征评价时要考虑不同化合物对风味的影响。如正丙醇、异丁醇和异戊醇是高级醇的代表物质, 赋予啤酒典型的“醇味”。正丙醇、异丁醇在啤酒中的浓度均低于其阈值的15 % ,对啤酒影响甚微,异戊醇的阈值70mg/ L ,浓度范围25~85mg/ L ,对啤酒有较大的影响。由于这些醇类混合物具有风味协同作用, 根据不同醇类对啤酒风味的影响比例大小及风味阈值, 可以得到啤酒“醇味”的风味强度值计算公式。由此建立的风味特征及相关化合物关系见表2 ,醇酯比关系见表3。
![]()
3. 2 判别不同啤酒的风味差别, 改善啤酒的风味稳定性和一致性
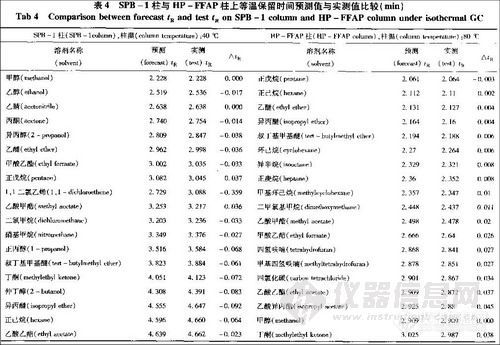
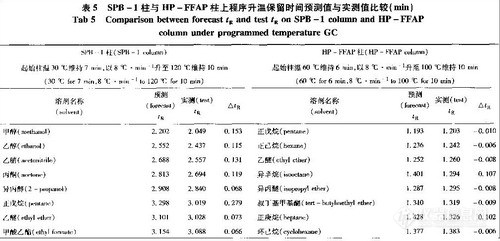
风味强度值结合风味物质的协同作用理论, 可以对不同时期、不同地点生产的啤酒比较风味差别,以区分其风味特征。风味差别度的评价方式见表4。
![]()
![]()
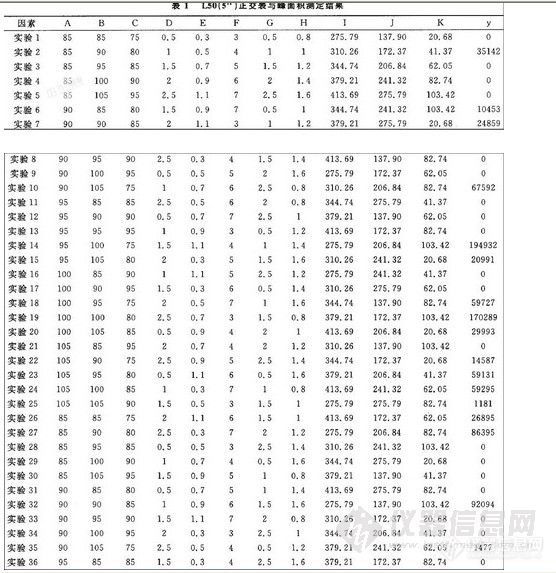
由表6 可见,以样品1 作对照,样品2 双乙酰味稍弱,酸败味稍强; 样品4 的酯香味较强; 样品5 的酯香味略强; 样品6 生青味略强。
利用风味强度值进行啤酒风味评价, 主要适用评价啤酒香气成分的主要骨架, 如高级醇、酯、双乙酰等有典型风味特征的物质。对于甜味、苦味、涩味等口感物质,由于不能进行定性定量分析,对其风味难以进行评价。因此,该方法与感官品评相结合,能对啤酒风味特征进行较全面的评价。
![]()