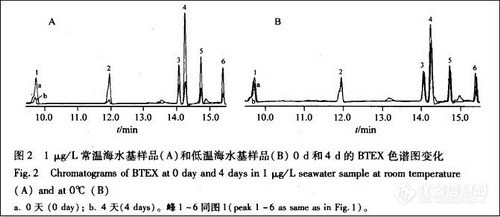
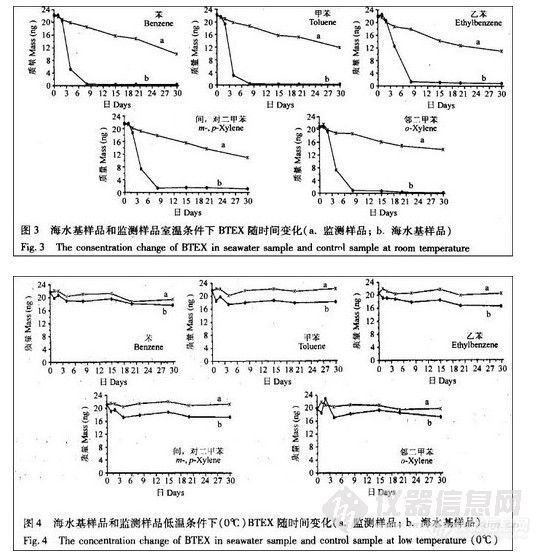
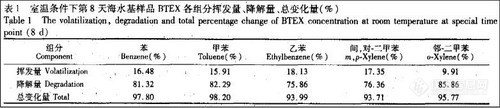
顶空气相色谱系列讲座(136)杭州市公共交通微环境中的苯系物污染
摘 要:
本文研究了杭州市公共交通流动微环境中苯系物的污染现状,监测了各种不同类型的
公交车、出租车内苯(BNZ)、甲苯(TOL)、乙苯(EBZ)、(邻-、间-、对-)二甲苯
(P-,m-,o-XY)、(1,3,5-、1,2,3-、1,2,4-)三甲苯( aTMB, bTMB, cTMB)、苯乙烯
(STR)的浓度。结果表明,流动微环境中苯系物总浓度为37.05-223.29μg/m3,其中
苯为8.81-34.15 μg/m3,暴露浓度远高于家庭、办公室等其他室内环境,污染较为严重。
本文由(1 浙江大学环境污染控制研究所,浙江大学环境与资源学院;(2 杭州职业技术学院;
(3 国家环保总局辐射环境监测技术中心;的李爽1,姚超英2,张平3,金苏君1,陈侠胜1,
沈学优1*合作完成,第一作者李爽是浙江大学硕士,主要研究方向为大气环境监测与分析。
本文摘自第四届全国环境化学学术大会文集下册。
前 言
苯系物是室内空气中重要的污染物之一,与白血病的高发有着较大的相关性。与室外空
气相比,室内空气污染更严重,人们的停留时间更长,苯系物对人体的影响更大。目前对苯
系物污染的研究主要针对在固定场所的,而缺少对流动微环境的研究报道。鉴于流动微环境
空间狭小、人流复杂、环境易受污染等特点,以及我国交通需求量急剧上升,人们使用交通
工具的频率和停留时间大大增加的现状,我们选择流动微环境作为研究对象。监测了杭州市
区公交车、出租车等公共交通工具乘客车厢10 种苯系物的浓度,综合评价其中苯系物的浓
度水平及其来源。丰富了室内污染监测的数据库,有助于我们了解城市中污染物的分布、迁
移转换规律和空气污染对人群健康的影响。
实 验
1 实验部分
1.1 采样点选择
本文选择公交车和出租车作为流动微环境的代表,详细监测了24辆公交车以及9 辆出
租车中苯系物的污染现状。采样时保持车辆正常运营时的状态。每辆车中同时采集两个平行
的空气样品,经分析后取平均值作为实验结果。采样时间2007年3~5月。
1.2 样品采集与测定
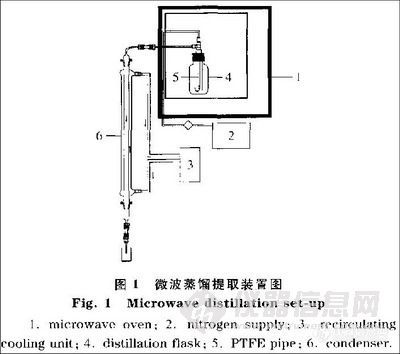
采样前将Tenax-TA 采样管在280℃下通N2处理1 h。采样时,保持车厢内正常运行时
的状况。采样时间为20 min,流量为0.5 L/min。测定时将采样管放入热解析器中,250 ℃
下热解析10 min,用N2作为载气将采集的样品完全带入
气相色谱中,进行分析。记录保留
时间和峰面积,以保留时间定性,峰面积定量,所有结果均换算到标准状态下。
2 结果与讨论
2.1 方法分析特征
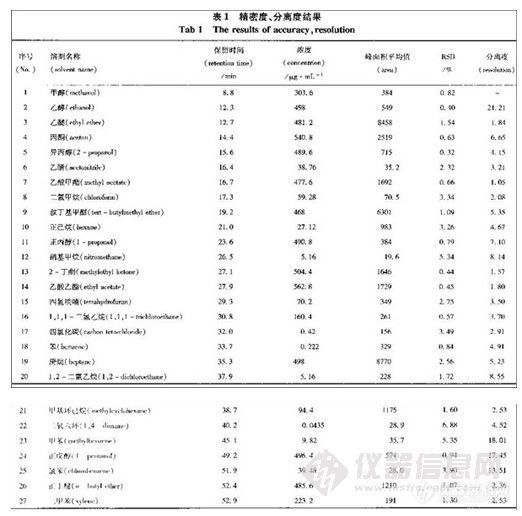
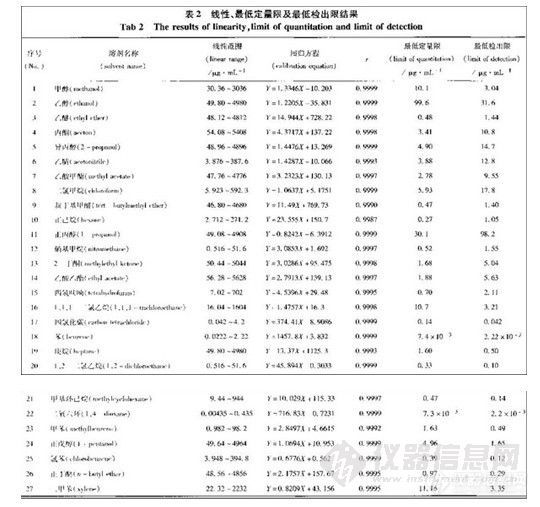
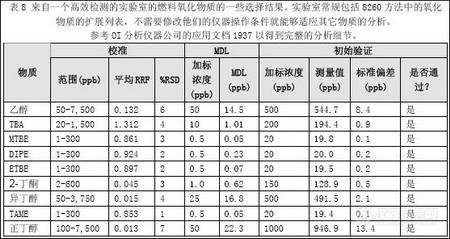
本实验方法可完全分离十种苯系物,方法检测限可达0.54ng/m3/10L以下。采用上述采
样和分析方法,10 种苯系物的回收率81%~101.8%,采样效率可达89.5%~99.3%。
2.2 苯系物污染水平
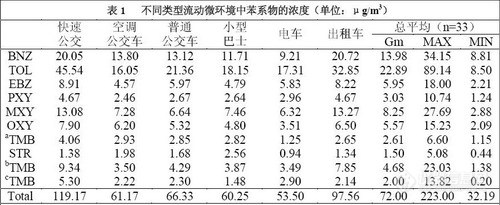
根据监测结果(见表1),杭州市公共交通流动微环境中苯系物总平均浓度为71.64μg/m-3,
其中甲苯浓度最高,为23.29μg./m-3,其次是苯,浓度为15.32μg./m-3。与课题组同期监
测所得的其他室内环境相比[1],流动微环境中苯系物总浓度远高于家庭、办公室等人们经常活
动的室内环境,其中对人体危害最大的苯的浓度高达其他室内环境的2.5 倍。
![]()
2.3 苯系物浓度的影响因素及其来源分析
苯系物的来源主要有室内源和室外源两大部分,其中室内源包括交通工具各种配件、材
料、内部装饰品以及液体燃料挥发进入流动微环境中;室外来源主要是附近车辆的尾气排放。
汽车尾气是流动微环境中苯系物的主要污染源。这也可以从不同苯系物的浓度比值(B/T/E/X)中
看出,在研究所得B/T/E/X 值为3:4:1:3,接近于Chiang 等的研究结果[2](汽车尾气中
B:T:E:X 值约为3:4:1:4),表明流动微环境中苯系物主要来自汽车尾气。
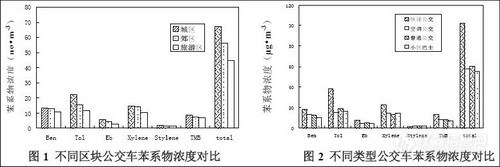
车辆行经区域苯系物的背景浓度对微环境中苯系物的含量有着极大的影响,公交车微环
境中苯系物浓度与采样过程中公交车经过的区块有很好的相关性,三个区块浓度由高到低分
别为城区>郊区>旅游区(见图1)。
公共交通工具的类型不同对苯系物的浓度有较大的影响,实验中,出租车内苯系物浓度
显著高于公交车中的浓度。不同类型公交车中苯系物浓度比较。不同类型公交车中苯系物总
浓度由高到低分别为快速公交>空调公交>普通公交>小区巴士(见图2)。
![]()
其中快速公交是杭州新开通的一类公交车,车辆较新,除了汽车尾气外还有车厢内的油
漆、地板革、布质座椅中的胶粘剂挥发,使得甲苯、乙苯和二甲苯的浓度大大增加;车辆窗
户紧闭,空气流通性差,车内物品挥发产生的苯系物不能及时得到扩散排斥,苯系物浓度高
于其他公交。
此外交通工具类型、所用的动力、行经的区域、通风状况等对微环境中苯系物的浓度均
有不同程度的影响,空调使用及乘客数量没有明显影响。
![]()