![]()
是这样,之前做一个样品,加内标的,除溶剂峰外,样品和内标峰形都对称,新柱子,DB1701的,刚用两个星期不到,结果工程师来维修过检测器,顺便对
气相大概讲解示范了一下,我们科是第一次做
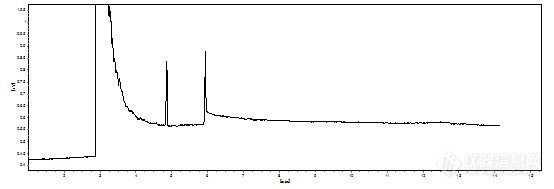
气相,结果隔一天在做,发现样品峰严重拖尾,可以说是趴着下来的,峰高很低,但内标就挺好,出峰顺序依次 溶剂,内标,样品
我程序升温,方法应该没问题,按照说明书和网上说的都大体排查了一遍,首先肯定不是进样量的问题,现在还算比较小的,隔膜清洗我没动过,但当时工程师动没动就不知道了,反正现在是开着的,进样口能清理的部分就是玻璃衬管,用乙醇超声20min,不放心,又用甲醇泡了一会,烘干,装上石英棉,装上,更换了进样垫,检测器的温度一直在270-300度,觉得没问题就没打算清理,毕竟比较麻烦,怕出更多问题,色谱柱连续几天都在老化,而且使用中温度不算低,结果这些做了之后还是样品峰拖尾
还有最后两步可以做,就是明天重装一下柱子(天美说的),再不行就截一段柱子(能不做就不做,短柱子啊,30米,截了就怕分不开了,而且怕我老师不理解,以前没接触过
气相,而且网上说的长度不一样,有说1,2圈的,有说1米的)
而且我还有个疑虑,就是关于玻璃衬管的清洗,天美维修工程师说用无水乙醇超声就行,但天美实验室的人说用硅烷化试剂冲一下,再用甲醇冲一下就行,我们这里的
气相老师说用洗液泡也行,要命的是我们这里没有硅烷化试剂,我只能在简单清洗而不能进行灭活处理,能用洗液泡吗?还是说用醇泡?
请大虾老师专家高手们给我出出主意吧!!!!!!
心情比较郁闷和沮丧,有点语无伦次,请谅解!