获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往

原文由 昂哩昂哩(jinxuan) 发表:
本人也咨询和测试过几家仪器供应商,较倾向于使用IC-ICP-MASS联用;一来不影响其他重金属测试,同时快速分离六价铬和三价铬测试。但所有仪器公司使用IC-ICP-MASS还是LC-ICP-MASS联用也好,关键看几点:色谱柱抓取条件能力(对样品溶液能达到延时分离六价铬时要求达到多少PH值);样品进样量(能同时支持其它17种重金属和六价铬、有机锡测试的样品溶液分配吗);分析高盐样品稳定性;高铬(或六价铬)浓度样品残留冲洗时间效果;
另询问下几个问题,1、使用的是IC配紫外检测器就行了吗?不是IC-UV-VIS联用吧?
2、使用的IC使用什么材料和直径的色谱柱?使用金属的进样针是否有影响稳定性?
3、现在你使用的方法需要提升柱温吗?用氨水调节样品PH值到多少?
4、没针进样量多少毫升样品溶液?
5、三价铬如何解决呢?直接总铬减去六价铬结果,还是三价铬和六价铬用色谱柱分离测试?
希望以上问题能与大家共同探讨下?谢谢大家帮助。
原文由 007爵士(jardz) 发表:原文由 shiyixue2000(shiyixue2000) 发表:原文由 rongdao(rongdao) 发表:原文由 007爵士(jardz) 发表:
我是这样考虑的。因六价铬的测试分3类,最严格是第二类液体样品为5ppb的限量。其他两类的限量为20ppb与200ppb。通过分析公司内部样品情况,发现属于第二类的样品不到2%,而且属于第一类20ppb的样品也不到2%,也就是约有95%以上样品只需要满足200ppb要求即可。那么,除了HPLC-ICPMS这么牛B仪器外,还有其他仪器其他方法能够满足这块要求吗?
尝试紫外分光光度计失败外,终于找到了离子色谱仪。(感谢某某检测机构资深技术经理深厚的友情,将该项的全程机密私授与我)也找到IC在其他行业测试六价铬的文献。立马联系厂家,去他们实验室实际考察做样情况。~~~~啪啦啪啦~~~带着2个样品(样品1经0.07M盐酸萃取的溶液;样品2经萃取后的溶液加入其他17种总金属,并且模拟实际样品中高含量的铝与锌元素)就冲到厂家实验室去了。原以为ICPMS都那么折腾的玩意儿,IC估计得难度更大,抱着怀疑且试试看的心态。经过氨水调PH值,上机测试(硫酸铵与氨水缓冲条件,二苯卡巴肼柱后衍生,紫外检测器测试)没想到不到半小时就解决了。太让我惊奇了~~~~不可能吧~~~~然后,让对方做了几针的重复性试验,加标试验,IDL摸索试验。惊人发现在复杂基质条件下的加标回收率1ppb浓度能达到90%左右,重复性是良好的,而且最低检出限能做到接近0.1ppb。因为萃取后的溶液处理过程只滴加了不到0.1ml的氨水。溶液最终的体积几乎没被稀释,也就是能够测试到样品中的浓度为5ppb(几乎是最严格的第二类限量值附近,该检出限附近的值还待考察,但是已经能够远远满足我第一三类的限量值)。与对方实验室技术经理沟通发现,该技术其实相当成熟,只是测试六价铬是运用其他行业,而且国际某大型检测机构(俺手里也有该机构使用IC检测六价铬完整报告),某出入境检验检疫局也早已采用该方法日常测试。而后交流了很多实际样品中出现的技术问题,均得到较为满意的答案。
(附上两个谱图,谱图1为0.1ppb标液;谱图2为复杂基质/复杂基质加标1ppb/标液1ppb)
谱图1:
谱图2:
你买了IC+UV了吗?测试效果怎么样?
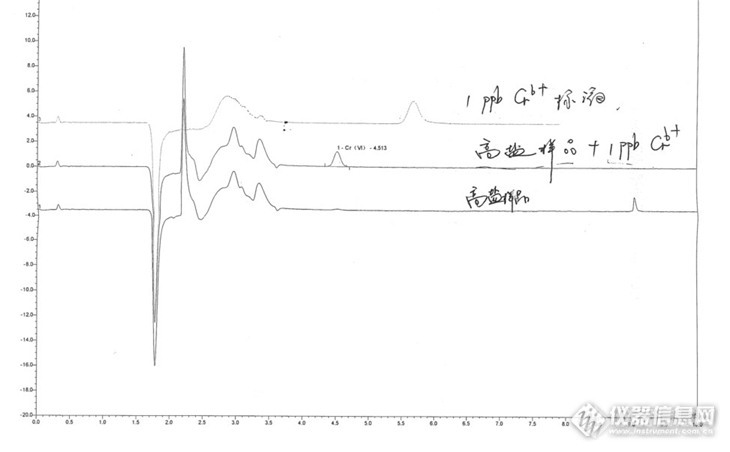
我看了第一张图的Cr6是在5.5分前出峰,第二张图中的最上面色谱图的Cr6是在5.5分后出峰,第二张图中的中间的色谱图的Cr6是在4.5分后出峰,这样的结果能相信吗?
第二张图中的最下面色谱图9分后出峰的是什么东西?能帮忙解释下吗?谢谢!
老板让我们也考察下什么仪器能做铬的形态。如果离子色谱能满足要求就最好了。如果不行,买回来摆在那里,老板要开骂的。我们只是企业用来自检。
同问:高盐样品、高盐加标、标准品Cr6+出峰时间都不一致,这如何定性呢?
======================================================
虽然帖子内小喷了A的ICPMS表现,但这几天却是在参加A公司的高级培训。
培训课程很紧,而且临近春节,所以有点忙,没能及时回复各位同行朋友,请谅解。
几位朋友问道IC保留时间有差异性问题,
1.因测试样品当天是采用之前曲线计算的,其标准溶液测试时间跟样品测试时间间隔了一周左右,保留时间略有差异。
2.可以确定的是,日常测试样品(非高盐样品)的加标出峰的保留时间与标准溶液的保留时间偏差不大。
3.高盐样品溶液对IC分离柱会产生很大影响,需要通过钠柱进行置换平衡,才能上机测试。(第二张图未经过钠柱进行上机,当时是考核最复杂样品条件对IC的影响,具体为什么保留时间往前偏移,回头具体解释。)
4.IC的定性跟HPLC类似,若需要判定是否为Cr VI,可以通过加标确定。
5.本文初衷是提醒同行们在六价铬项目上除了LC-ICPMS外,还可以使用IC-UV。而涉及IC-UV测试过程中还有很多细节的技术问题未详尽描述,请见谅,我将陆续完善,也请朋友们多多交流。
性急或打算考核厂家仪器的朋友,可先私信我。
原文由 xuquanhui(xuquanhui) 发表:
[关于保留时间差异的问题,看到很多版友提问,楼主也没有完全说明白。我觉得可以从图上分析:
1,4分钟以前的那一堆峰是系统峰以及样品中的基体造成的。IC-UV的测定原理和比色法是一样的,只要能和二苯卡巴肼反应生产有光吸收的产物,或者基体本身有光吸收的,都会在色谱图上造成“峰”。只要这些峰的位置固定,而且能证实不是六价铬,一般是不管的。
2,六价铬的峰应该是出在4~6分钟之间的。楼主几张图中的六价铬峰保留时间确实相差很大,主要原因应该是样品的离子强度和标准品相差很大。样品中含有70mmol/L相当于两千多ppm的Cl离子,会影响六价铬在柱子上的保留行为,保留时间有偏差是正常的。如果你觉得偏差大到不能接受,也有一个办法,就是过Ag柱,把Cl去掉,只是Ag柱很贵,又是一次性的,可能他们舍不得;或者担心影响六价铬的回收率,因为铬酸银也是不溶,得具体试试才知道效果如何。
3,楼主说道样品和标准品隔了一周,所以保留时间不同。我倒觉得不是这个原因,按说只要保证了流动相一致、柱子一致、衍生的条件(特别是酸度和温度,这是比较苛刻的)一致、基体基本匹配,一个月甚至半年都变化不大的。
4,9分钟左右的那个峰,不像六价铬,估计是一个高价态的过度金属离子,或者是一个金属的氢氧化物胶体之类的东西跑出来了,只要有光吸收,色谱图上就有用反应。当碰到含较高的三价铬、铁、锰、钒之类的样品,往往会有这些金属的氢氧化物沉积在柱子里,不知道什么时候跑出来,小的产生毛刺,大的就像一个峰。
5,楼主你说的过钠柱,是想把样品中的阳离子除掉吗?除掉阳离子是有好处,不会再有4的担心了。但其实影响六价铬保留时间最大的还是阴离子,因为你在IC上用的是阴离子交换柱,六价铬也是以阴离子形式存在的,我认为过Ag柱的效果会比过Na柱好。两个柱都过一遍当然更好,但要考虑成本和回收率啊。
原文由 xuquanhui(xuquanhui) 发表:原文由 007爵士(jardz) 发表:原文由 shiyixue2000(shiyixue2000) 发表:原文由 rongdao(rongdao) 发表:原文由 007爵士(jardz) 发表:
我是这样考虑的。因六价铬的测试分3类,最严格是第二类液体样品为5ppb的限量。其他两类的限量为20ppb与200ppb。通过分析公司内部样品情况,发现属于第二类的样品不到2%,而且属于第一类20ppb的样品也不到2%,也就是约有95%以上样品只需要满足200ppb要求即可。那么,除了HPLC-ICPMS这么牛B仪器外,还有其他仪器其他方法能够满足这块要求吗?
尝试紫外分光光度计失败外,终于找到了离子色谱仪。(感谢某某检测机构资深技术经理深厚的友情,将该项的全程机密私授与我)也找到IC在其他行业测试六价铬的文献。立马联系厂家,去他们实验室实际考察做样情况。~~~~啪啦啪啦~~~带着2个样品(样品1经0.07M盐酸萃取的溶液;样品2经萃取后的溶液加入其他17种总金属,并且模拟实际样品中高含量的铝与锌元素)就冲到厂家实验室去了。原以为ICPMS都那么折腾的玩意儿,IC估计得难度更大,抱着怀疑且试试看的心态。经过氨水调PH值,上机测试(硫酸铵与氨水缓冲条件,二苯卡巴肼柱后衍生,紫外检测器测试)没想到不到半小时就解决了。太让我惊奇了~~~~不可能吧~~~~然后,让对方做了几针的重复性试验,加标试验,IDL摸索试验。惊人发现在复杂基质条件下的加标回收率1ppb浓度能达到90%左右,重复性是良好的,而且最低检出限能做到接近0.1ppb。因为萃取后的溶液处理过程只滴加了不到0.1ml的氨水。溶液最终的体积几乎没被稀释,也就是能够测试到样品中的浓度为5ppb(几乎是最严格的第二类限量值附近,该检出限附近的值还待考察,但是已经能够远远满足我第一三类的限量值)。与对方实验室技术经理沟通发现,该技术其实相当成熟,只是测试六价铬是运用其他行业,而且国际某大型检测机构(俺手里也有该机构使用IC检测六价铬完整报告),某出入境检验检疫局也早已采用该方法日常测试。而后交流了很多实际样品中出现的技术问题,均得到较为满意的答案。
(附上两个谱图,谱图1为0.1ppb标液;谱图2为复杂基质/复杂基质加标1ppb/标液1ppb)
谱图1:
谱图2:
你买了IC+UV了吗?测试效果怎么样?
我看了第一张图的Cr6是在5.5分前出峰,第二张图中的最上面色谱图的Cr6是在5.5分后出峰,第二张图中的中间的色谱图的Cr6是在4.5分后出峰,这样的结果能相信吗?
第二张图中的最下面色谱图9分后出峰的是什么东西?能帮忙解释下吗?谢谢!
老板让我们也考察下什么仪器能做铬的形态。如果离子色谱能满足要求就最好了。如果不行,买回来摆在那里,老板要开骂的。我们只是企业用来自检。
同问:高盐样品、高盐加标、标准品Cr6+出峰时间都不一致,这如何定性呢?
======================================================
虽然帖子内小喷了A的ICPMS表现,但这几天却是在参加A公司的高级培训。
培训课程很紧,而且临近春节,所以有点忙,没能及时回复各位同行朋友,请谅解。
几位朋友问道IC保留时间有差异性问题,
1.因测试样品当天是采用之前曲线计算的,其标准溶液测试时间跟样品测试时间间隔了一周左右,保留时间略有差异。
2.可以确定的是,日常测试样品(非高盐样品)的加标出峰的保留时间与标准溶液的保留时间偏差不大。
3.高盐样品溶液对IC分离柱会产生很大影响,需要通过钠柱进行置换平衡,才能上机测试。(第二张图未经过钠柱进行上机,当时是考核最复杂样品条件对IC的影响,具体为什么保留时间往前偏移,回头具体解释。)
4.IC的定性跟HPLC类似,若需要判定是否为Cr VI,可以通过加标确定。
5.本文初衷是提醒同行们在六价铬项目上除了LC-ICPMS外,还可以使用IC-UV。而涉及IC-UV测试过程中还有很多细节的技术问题未详尽描述,请见谅,我将陆续完善,也请朋友们多多交流。
性急或打算考核厂家仪器的朋友,可先私信我。
关于保留时间差异的问题,看到很多版友提问,楼主也没有完全说明白。我觉得可以从图上分析:
1,4分钟以前的那一堆峰是系统峰以及样品中的基体造成的。IC-UV的测定原理和比色法是一样的,只要能和二苯卡巴肼反应生产有光吸收的产物,或者基体本身有光吸收的,都会在色谱图上造成“峰”。只要这些峰的位置固定,而且能证实不是六价铬,一般是不管的。
2,六价铬的峰应该是出在4~6分钟之间的。楼主几张图中的六价铬峰保留时间确实相差很大,主要原因应该是样品的离子强度和标准品相差很大。样品中含有70mmol/L相当于两千多ppm的Cl离子,会影响六价铬在柱子上的保留行为,保留时间有偏差是正常的。如果你觉得偏差大到不能接受,也有一个办法,就是过Ag柱,把Cl去掉,只是Ag柱很贵,又是一次性的,可能他们舍不得;或者担心影响六价铬的回收率,因为铬酸银也是不溶,得具体试试才知道效果如何。
3,楼主说道样品和标准品隔了一周,所以保留时间不同。我倒觉得不是这个原因,按说只要保证了流动相一致、柱子一致、衍生的条件(特别是酸度和温度,这是比较苛刻的)一致、基体基本匹配,一个月甚至半年都变化不大的。
4,9分钟左右的那个峰,不像六价铬,估计是一个高价态的过度金属离子,或者是一个金属的氢氧化物胶体之类的东西跑出来了,只要有光吸收,色谱图上就有用反应。当碰到含较高的三价铬、铁、锰、钒之类的样品,往往会有这些金属的氢氧化物沉积在柱子里,不知道什么时候跑出来,小的产生毛刺,大的就像一个峰。
5,楼主你说的过钠柱,是想把样品中的阳离子除掉吗?除掉阳离子是有好处,不会再有4的担心了。但其实影响六价铬保留时间最大的还是阴离子,因为你在IC上用的是阴离子交换柱,六价铬也是以阴离子形式存在的,我认为过Ag柱的效果会比过Na柱好。两个柱都过一遍当然更好,但要考虑成本和回收率啊。
原文由 007爵士(jardz) 发表:原文由 xuquanhui(xuquanhui) 发表:
[关于保留时间差异的问题,看到很多版友提问,楼主也没有完全说明白。我觉得可以从图上分析:
1,4分钟以前的那一堆峰是系统峰以及样品中的基体造成的。IC-UV的测定原理和比色法是一样的,只要能和二苯卡巴肼反应生产有光吸收的产物,或者基体本身有光吸收的,都会在色谱图上造成“峰”。只要这些峰的位置固定,而且能证实不是六价铬,一般是不管的。
2,六价铬的峰应该是出在4~6分钟之间的。楼主几张图中的六价铬峰保留时间确实相差很大,主要原因应该是样品的离子强度和标准品相差很大。样品中含有70mmol/L相当于两千多ppm的Cl离子,会影响六价铬在柱子上的保留行为,保留时间有偏差是正常的。如果你觉得偏差大到不能接受,也有一个办法,就是过Ag柱,把Cl去掉,只是Ag柱很贵,又是一次性的,可能他们舍不得;或者担心影响六价铬的回收率,因为铬酸银也是不溶,得具体试试才知道效果如何。
3,楼主说道样品和标准品隔了一周,所以保留时间不同。我倒觉得不是这个原因,按说只要保证了流动相一致、柱子一致、衍生的条件(特别是酸度和温度,这是比较苛刻的)一致、基体基本匹配,一个月甚至半年都变化不大的。
4,9分钟左右的那个峰,不像六价铬,估计是一个高价态的过度金属离子,或者是一个金属的氢氧化物胶体之类的东西跑出来了,只要有光吸收,色谱图上就有用反应。当碰到含较高的三价铬、铁、锰、钒之类的样品,往往会有这些金属的氢氧化物沉积在柱子里,不知道什么时候跑出来,小的产生毛刺,大的就像一个峰。
5,楼主你说的过钠柱,是想把样品中的阳离子除掉吗?除掉阳离子是有好处,不会再有4的担心了。但其实影响六价铬保留时间最大的还是阴离子,因为你在IC上用的是阴离子交换柱,六价铬也是以阴离子形式存在的,我认为过Ag柱的效果会比过Na柱好。两个柱都过一遍当然更好,但要考虑成本和回收率啊。
=============================================
仁兄果然高人呀!
你另开的专门帖子http://bbs.instrument.com.cn/shtml/20130217/4570309/index_1.shtml,精品呀,内容甚广,我定会好好研读。
祝你仪器大卖呀!恭喜发财!!!
ICPMS我们还是会买的。哈哈,我倒是希望你多来几次, 多讲点专业信息,定当受益匪浅呀。。。
原文由 unknown8888(unknown8888) 发表:原文由 007爵士(jardz) 发表:原文由 thermoczq(thermoczq) 发表:
...这个肯定是T公司的ICPMS+D公司的IC啦,不过现在都是T公司的。看上次好像可以做,但感觉有点过度宣传IC,忽略了大头ICPMS,本末倒置了
EN71-3修订F版中有提及使用IC-UVis,以及IC-ICPMS测试方法。我写这篇文章目的是提醒各位朋友在六价铬项目上不是只有HPLC-ICPMS才具备相应的测试能力,不要过分相信ICPMS仪器商的胡吹乱侃。需要结合自身条件(日常测试样品是否真的很大比例的样品需要满足第二类最低的限量要求?除了大费用购买HPLC-ICPMS外,是否核算过日常的测试成本?)。。。找到适合自己的才是王道。。。。
EN71-3:2013要做第二类限值的六价铬,那必须保证:
1、仪器整个体系足够干净(那么仪器在做二类测试样品前,必须冲洗足够长的时间,时间成本必须考虑);
2、二类测试要求测试过程、前处理过程用到的水、试剂、标准品都必须具有最高的纯度(二类六价铬的标准曲线要达到PPT级,哪怕ppt级别的六价铬污染都会对二类六价铬定量造成很大干扰。试剂成本必须考虑);
3、即便六价铬标准溶液在ppt级别上有相对较好的相应和平滑的基线,但实际样品基体的复杂性,会给PPT级别的六价铬的分离造成很严重的影响(基体的复杂会给PPT级的六价铬的基线带来非常大的波动,不可能要求所有第二类测试样品都必须是简单的基体环境)。
所以,无论是用LC-ICP-MS还是用IC来测试,对于第一类和第三类六价铬来讲,两种仪器的灵敏度和重现性都能很好的满足要求了,而对于测限值非常低的第二类六价铬,影响因素最大的不是仪器性能的差别,反而是尽可能的防止各个测试环节中六价铬污染的引入。
可以说:二类的六价铬测试,要想取得比较好的定量结果和重现性,那是在一种“简单的样品基质,在非常非常干净的试剂,标品,仪器体系的测试环境中”才有可能达到的理想状态。
我们不能简单的下结论:LC-ICPMS能做第三类,IC做不了第三类。