维权声明:本文为jieqian1211原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。
很不幸,盲样考核又到了兄弟手上,还好本次测定的是奶粉中的锌,锌还算比较稳定的,虽然污染源较多,但是加以控制还是可以克服的。对待盲样考核,我一直比较慎重,每次都要制定出工作思路,这次也不例外。
目前火焰
原子吸收测锌主要存在的问题就是污染,因为锌的灵敏度较高,1.6ppm的锌在安捷伦AA24oFS型
原子吸收上其吸光度大约为0.7,这超过了我所制定的吸光度控制在0.5以内的预期,因此初步考虑要降低其吸光度。
顺便提一句,我们这边系统内的很多人曾经都问过我为什么锌那么不好测啊,每次线性都做不好,问我有什么高招。我就问其标准系列最高点多少啊,最高点吸光度多少啊,样品空白吸光度多少啊?他们给出的回答基本上都千遍一律,最高点大都到了2.0ppm,吸光度也都在0.5以上,空白的吸光度有些都到了0.03以上。我就说你们想办法降低灵敏度或者降低最高点浓度,把所有点的吸光度都控制在0.5以内(其实基层实验室的环境远远无法采取降低标准系列浓度来克服这个问题)所以他们最终采取的办法就是偏转燃烧头,严格控制空白吸光度在0.01以下。
而我这次也面临这样的问题,因为根据以往做奶粉的经验,处理后的奶粉中锌含量一般都接近1.0ppm,考虑到称样量太小对结果的影响,我们还只能称取0.5g以上的样品,而且尽量避免逐级稀释,因为每一次稀释都引入了误差。所以最终我们也只选择了偏转燃烧头的办法。
分析的基础问题解决了,那就要来肢解分析步骤。
第一,前处理。手上可用的工具:微波消解仪,电热板,马弗炉。本次样品量在35g左右,完全够用,所以三个方法都试一试。但主要以微波消解和电热板湿法消解为主;
第二步,预实验,确定取样量和标准系列的范围。根据微波消解本身的特点,称样量最好控制在0.5g左右,为了尽量减少酸的用量电热板湿法消解称样量也控制1.0g左右,干法控制在2.0g左右。
第三步,质控。要用类似基质的质控,还好奶粉质控比较好买。根据经验我们选择了奶粉生物成分的质控样,尽量接近盲样的值。
以上思路确定。那就具体操作吧。
第一步,取样。干法取样在2.0g左右,微波取样在0.5g左右,湿法取样在1.0g左右。
![]()
第二步,消解。有三种方法。湿法和干法所用器皿均为石英烧杯,其可耐1000度的高温,在马弗炉里完全可以承受得住。
消解条件:微波:
![]()
湿法:加入12ml混酸(高氯酸+硝酸=1+4),过夜。第二天于电热板上180度消解至澄清透明。
干法:加入2ml硝酸,电路上小火碳化,然后转至大火至无烟,放入陶瓷马弗炉中550度保持5.0h。
![]()
全部消解完毕后,用去离子水定容,干法消解的用1%硝酸定容。微波消解定容至25ml,干法和湿法消解定容至50ml。定容过程中用百分之一天平对定容体积进行监视。
![]()
第三步,上机检测。
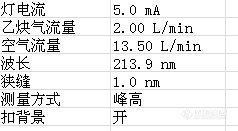
1.仪器条件:
![]()
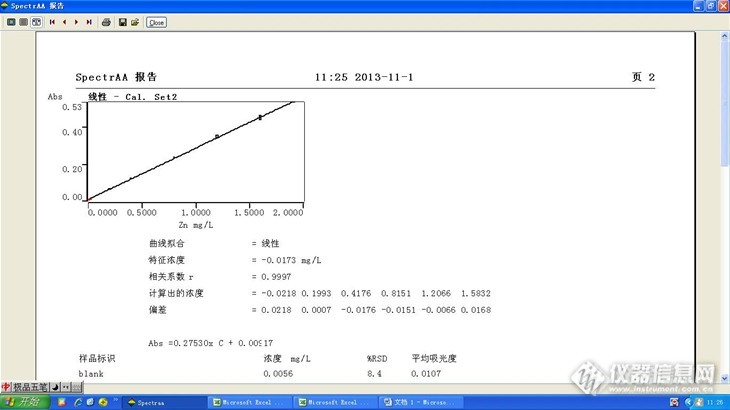
标准系列及标准曲线:
![]()
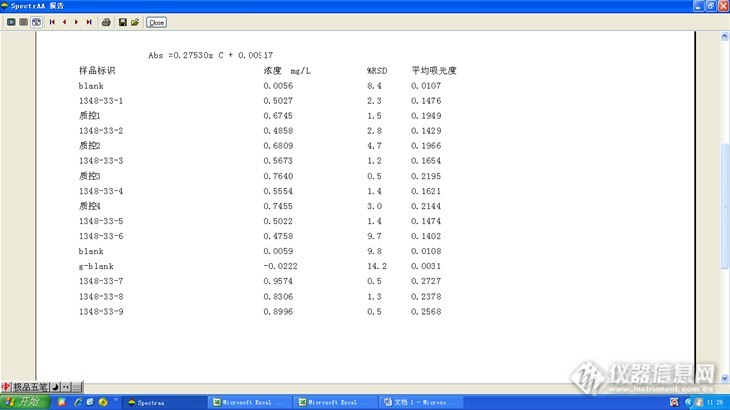
分析原始浓度:
![]()
第四步,结果分析。
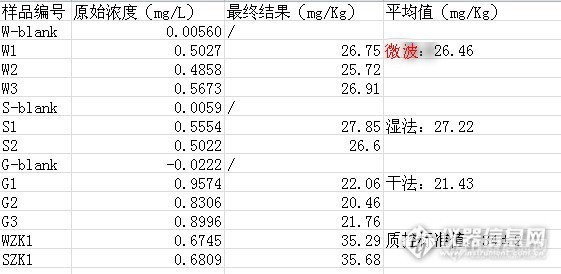
由于1348-33-6标准偏差较大,予以排除。再根据重量计算结果如下表:
![]()
从结果可以看出湿法和微波消解的结果差不多,而且质控都在参考值范围(34±2mg/Kg)之内,干法消解结果与湿法和微波消解结果相差较大,不可信,考虑到可能碳化灰化阶段损失所致。
注意事项 :为了验证吸光度在0.5以上对标准曲线的影响,我们做了一个小实验。
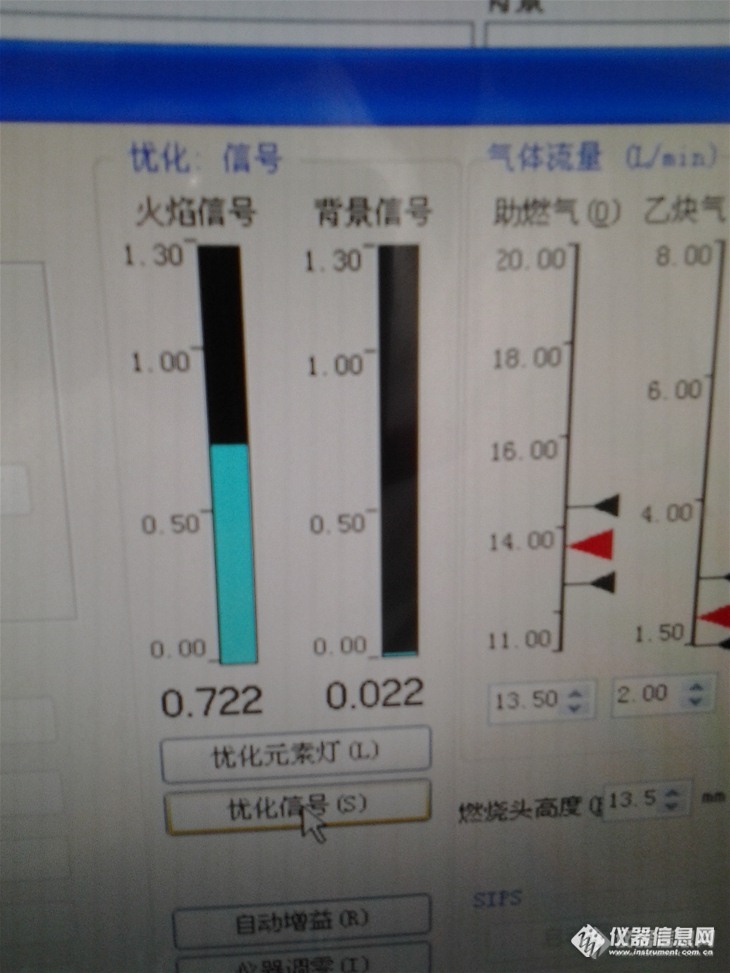
以 1.6ppm的标准点进样考察吸光度:发现吸光度在0.7以上。如下图:
![]()
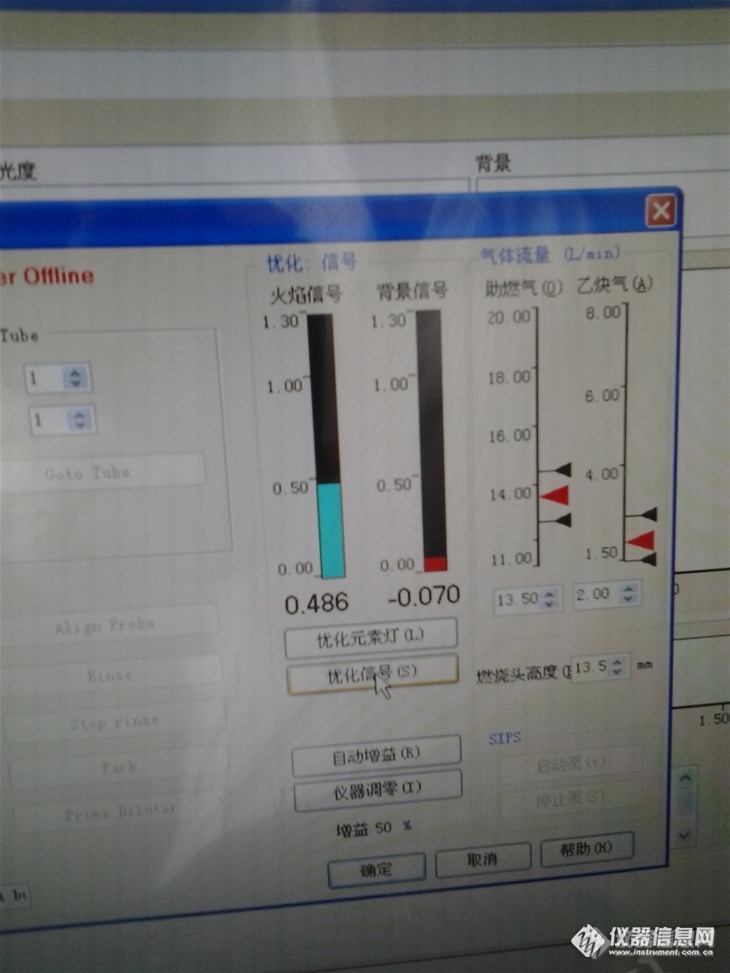
偏转燃烧头以后的情况:
![]()
![]()
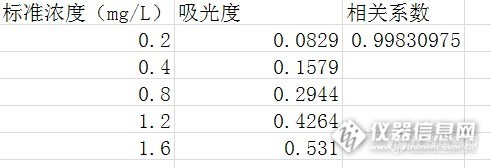
然后保持吸光度在0.5附近,进行标准系列测定,发现在最高点在1.6ppm,吸光度为0.531,结果标准系列的相关系数才0.998.如下图
![]()
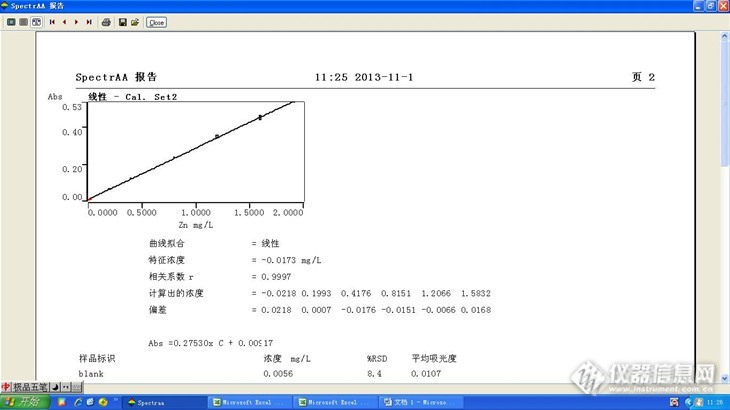
然后继续偏转吸光度,确保最高点吸光度在0.5以下,同一套标准系列,其相关系数达到0.9997。
![]()
由以上小实验可以发现,
原子吸收进行样品检测的时候为了获得良好的线性,最好保持最高点吸光度在0.5以内。