“色”路蹒跚,即鹿无虞,某品种有关物质方法学流动相摸索。
因暂时没有CERI L-column色谱柱,挑选了以前常用的色谱柱,进行了流动相的摸索,日产某品牌因理论板数较低(小于2000),故没有采用,选用月旭的色谱柱,一切皆好,试验完成后,把工艺处方摸索的样品都测试完成后,发现是用的苯基柱,虽说都是反相色谱柱,和标准不一致,要验证的,真是即鹿无虞啊!
有关物质避光操作。照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定。色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂(CERI L-column ODS,4.6mm×250mm,5μm或同等效能的色谱柱);流动相A为pH3.8醋酸钠缓冲液[取稀醋酸10ml,加水稀释至1000ml,取此溶液800ml,加醋酸钠溶液(取醋酸钠试液1ml,加水稀释至100ml)100ml,混匀],流动相B为乙腈,按下表进行梯度洗脱: 时间(分钟) | 0 | 20 | 40 | 70 | 71 | 80 |
流动相A(%) | 60 | 60 | 30 | 30 | 60 | 60 |
流动相B(%) | 40 | 40 | 70 | 70 | 40 | 40 |
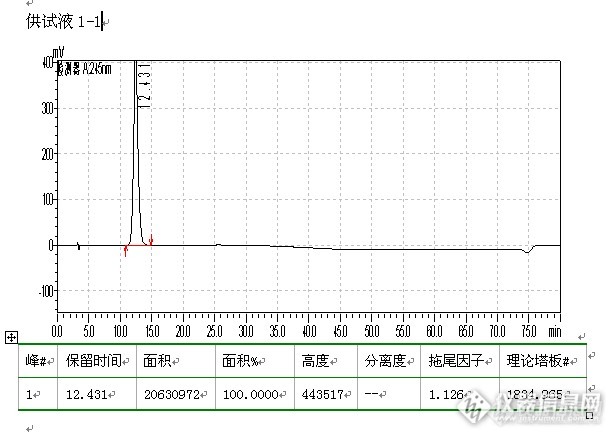
柱温为40℃,检测波长245nm。取苯甲酸乙酯约0.2g,置50ml量瓶中,加乙腈-水(3:2)溶解并稀释至刻度,混匀。取此溶液和供试品溶液各5ml置同一50ml量瓶中,用乙腈-水(3:2)稀释至刻度,摇匀,作为分离度测试溶液,量取10μl注入液相色谱仪,调整色谱系统,使匹伐他汀的保留时间约为23分钟,取对照溶液重复进样6次,主峰面积的相对保留时间偏差应小于9%,匹伐他汀与苯甲酸乙酯峰之间的分离度不小于5。测定法 取本品5片,置50ml量瓶中,加水20ml,振摇10分钟至片剂崩解,再超声5分钟,加乙腈20ml,振摇10分钟,加乙腈稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液;精密量取供试品溶液适量,加乙腈-水(3:2)稀释制成每1ml中约含匹伐他汀钙0.2μg的溶液,作为对照溶液。量取对照溶液10μl,注入液相色谱仪,调节检测灵敏度,使匹伐他汀的峰高约为记录仪器量程的5~10%,再精密量取供试品和对照溶液各10μl,注入液相色谱仪,记录色谱图。供试品溶液色谱图中除溶剂峰与辅料峰外,差向异构体(相对保留时间约为1.1)、内酯化合物(相对保留时间为1.7)和其他未知杂质的峰面积分别不得过对照溶液主峰面积的7.5倍(0.75%)、5倍(0.5%)和1倍(0.1%),杂质总量不得过对照溶液主峰面积的20倍(2.0%)。色谱柱信息:1.日产的C18色谱柱(都是大家最常用和常见品牌,未写出,主要怕麻烦)。2.月旭的C18色谱柱,在使用的过程中,操作人员弄成月旭的苯基柱。苯基柱信息:苯基柱:SN:241301633, LN:2401.11 日产C18色谱图:![]()
上图显示,理论板数较低,只有1834,虽说都是新柱子,也可能是流动相不适合该色谱柱,保留时间较短,要达到23分钟,调整流动相比例,估计理论板数会更低,故没有再折腾。
![]()
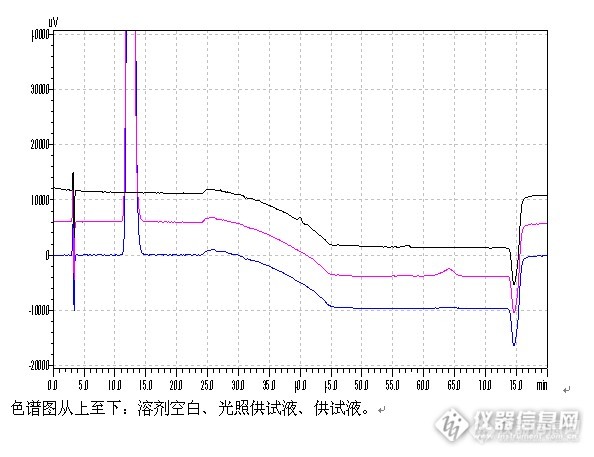
上图显示,其基线重现性良好,显示了新柱子的基本性能。
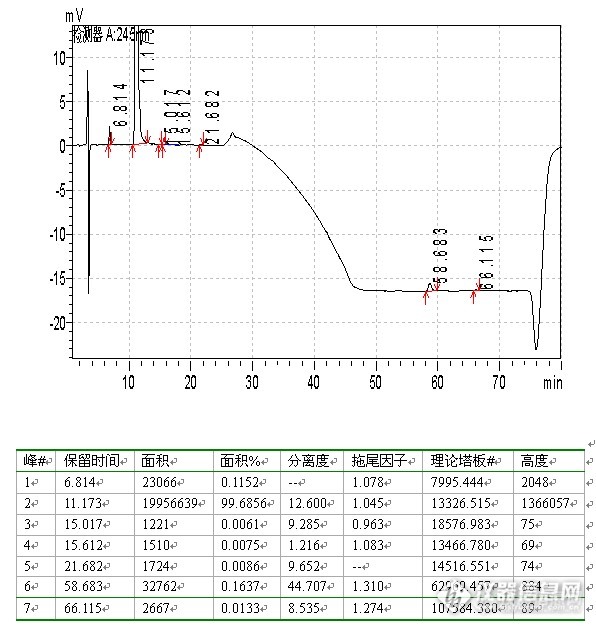
月旭苯基柱色谱图:
![]() 上图显示,理论板数较高,超过了10000,分离度和拖尾因子均较好,调整流动相比例,应该是有可能的。调整流动相比例色谱图:
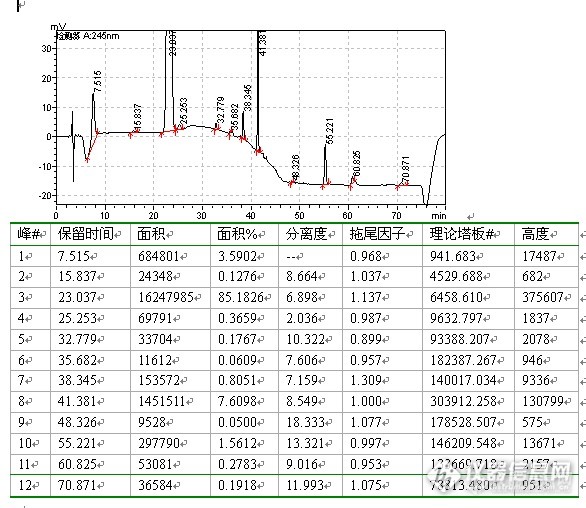
上图显示,理论板数较高,超过了10000,分离度和拖尾因子均较好,调整流动相比例,应该是有可能的。调整流动相比例色谱图:![]()
上图显示,分离度,拖尾因子,理论板数均好,大功告成(高兴的较早哈,呵呵。。。)
下图为放大显示:
![]()
总结,做试验是个细致活,在没有充足的条件下,创造条件,是好事,但是要注意细节(注意看标签,不要看到月旭的就想到了是C18),不然会走弯路的,我现在要进行色谱柱验证了,和C18进行对比。