获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往
表1

表2

表3

表4 Pb标系及强度
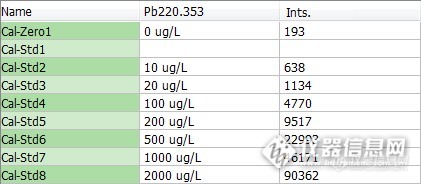
表5 Cd标系及强度
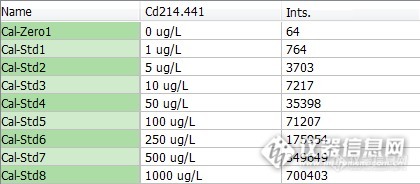
表6 Cr标系及强度

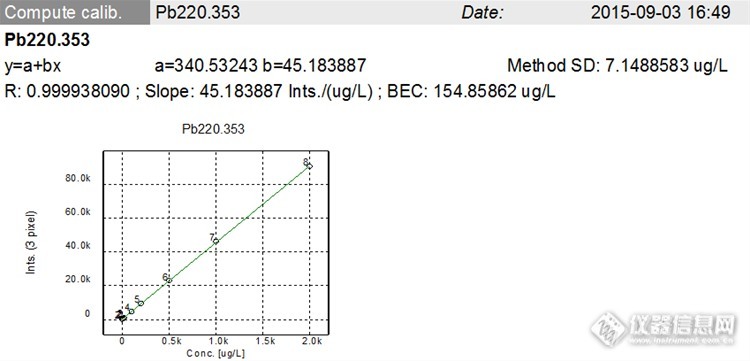

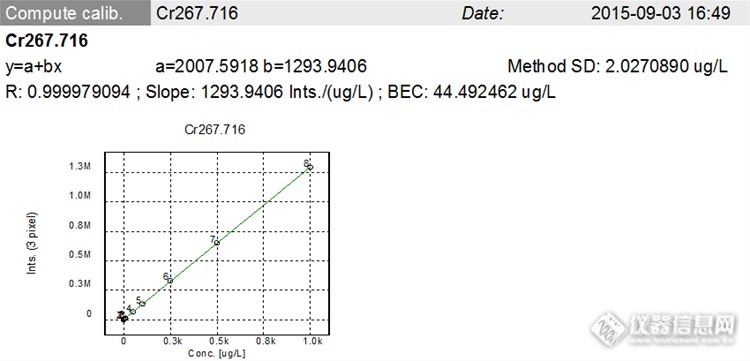
表7 回归方程及相关系数
元素 | 一次回归方程 | R |
Pb | y=340.53243+45.183887x | 0.999938 |
Cd | y=365.57285+699.93198x | 0.999997 |
Cr | y=2007.5918+1293.9406x | 0.999979 |
2.3.5 检出限LOD及定量下限LOQ测定(由仪器自动计算得出)
表8 检出限

2.3.6 样品测量
样品扣空白后中各元素测定数据记录与处理
表9 样品测定数据及处理

上表9,土壤水分含量测定以及土壤pH测定,分别参考GB/T17141-1997附录A 与NYT 1121.2-2006 土壤检测 第2部分:土壤pH的测定
土壤中Pb、Cd、Cr三种元素含量的各级标准如下表所示:
表10 土壤环境质量国家标准 (GB 15618-1995) 单位:mg/kg
级别 | 一级标准 | 二级标准 | 三级标准 | ||
pH值 | 自然背景 | <6.5 | 6.5-7.5 | >7.5 | >6.5 |
Pb≤ | 35 | 250 | 300 | 350 | 500 |
Cd≤ | 0.2 | 0.3 | 0.3 | 0.6 | 1.0 |
Cr≤(旱地) | 90 | 150 | 200 | 250 | 300 |
对比国家标准可知,对于Pb元素第4号样符合二级标准,其余样品均符合一级标准;对于Cd元素第4、7、10、12、16、18、20号样符合二级标准,其余样品均符合一级标准;对于Cr元素所有样品均符合一级标准
4 讨论
4.1 对比ICP法与石墨炉原子吸收法(ZEEnit700)的检出限
表11
| 元素 | ICP | GF-AAS | ICP能否替代GF-AAS | ||||
浓度范围 μg/L | 检出限 μg/L | 定量下限 μg/L | 浓度范围 μg/L | 检出限 μg/L | 定量下限 μg/L | ||
| Pb | 0、10、20、100、200、500、1000、2000 | 1.9 | 5.5 | 0、4、10、20、30、40 | 0.2 | 0.6 | ? |
| Cd | 0、1、5、10、50、100、250、500、1000 | 0.11 | 0.32 | 0、1、2、3、4 | 0.2 | 0.6 | ok |
| Cr | 0、1、5、10、50、100、250、500、1000 | 0.19 | 0.55 | 0、2、4、8、12、16 | 0.9 | 2.7 | ok |
由表11可以得出:
1.ICP的线性范围明显高于GF-AAS。
2.Cd与Cr的ICP法测定检出限与定量下限均低于GF-AAS,对于这两种元素的测定完全可以用ICP法替代。
3.ICP测定Pb的检出限比GF-AAS高出近十倍,对于土壤这类的Pb含量高的样品来说其测定可以用ICP法替代,但是对于超低含量的Pb元素测定石墨炉原子吸收法要优于ICP法。
4.从Pb的ICP定量下限可以看出,用ICP测Pb元素时,Pb的标系最低点不能低于5.5μg/L。
5.从三元素的定量下限可以看出,三元素的曲线最低点的设置还算合理
4.2 加标回收率
表12
元素 | 本底值(mg/kg) | 加标量(mg/kg) | 测定值(mg/kg) | 回收率(%) |
| Pb | 15.3 | 300 | 299.0 | 94.6 |
19.6 | 300 | 286.1 | 88.8 | |
| Cd | 0.084 | 0.3 | 0.292 | 69.4 |
0.241 | 0.3 | 0.433 | 64.3 | |
| Cr | 38.5 | 200 | 247.6 | 104.6 |
37.2 | 200 | 236.2 | 99.5 |
从上表可以看出Cd的回收率明显偏低,原因还需进一步查找。
4.3 平行双样相对偏差
表13
元素 | 样品1 (μg/L) | 平行样1 (μg/L) | 平行样品相对偏差(%) |
Pb | 190 | 187.5 | 1.32 |
Cd | 1.716 | 1.707 | 0.52 |
Cr | 296.5 | 300.8 | 1.44 |
从平行双样相对偏差可以看出本次消化以及仪器的稳定性良好
4.4 本实验室ICP配备自动进样器,整个进样管路较长,测定基体复杂的样品时最好在软件界面设置延长样品间的清洗时间,以尽量减少样品间的交叉污染。在完成曲线测定后,也最好紧接着增加一步清洗进样系统,以尽量减少甚至消除曲线最高浓度点标液残留对后续样品测定的干扰。在完成所有样品测定后,先保持吸液管在冲洗液中冲洗2分钟,再将进样管插入超纯水中冲洗2分钟,最后将样品管从超纯水中取出,吸入空气,等雾化室中的雾气消失时,管路中的液体即被排空,可关闭火焰。保证熄火前管路中没有液体残留。如果管路中有试剂残留特别是酸性的样液残留时,会造成自动进样器上的橡胶蠕动管提前老化。
4.5 点火前先确认CCD检测器冷却至-10℃,冷却循环水状态正常,排风正常,再吹扫雾化室,吹扫一次60秒,可以连续吹扫两次再点火
4.6 Excel公式计算注意事项:
按照GB/T17141-1997中的公式人工计算Pb,Cd含量时,会人为的转化单位,所以按照该标准的公式计算是没有问题的。但是如果像我一样习惯Excel处理数据的就要注意了,在编辑公式时要人为的除以转换系数1000。即: W=(c-c0)*V/(m*(1-f))/1000 (各个参数的单位与标准一样)
5 结论
本实验采用EPA3050B消解技术,运用电感耦合等离子体发射光谱法(ICP-OES)测定土壤中的Pb、Cd、Cr含量。实验表明:本批次的土壤样品中Pb的含量范围为12.1-35.7mg/kg,加标回收率为88.8%-96.4%;Cd的含量范围为0.055-0.536mg/kg,加标回收率为64.3%-69.4%;Cr的含量范围为28.2-44.8mg/kg,加标回收率为99.5%-104.6%。各元素的平行双样相对偏差均 <2%。ICP-OES法测定土壤中重金属的含量,具有线性范围宽,精密度高,检出限低,分析速度快并且能多元素同时测定等优点,但也有仪器昂贵、氩气消耗量大的缺点,对于有条件的实验室可以推广应用于土壤中重金属的检测。

原文由 henkyq(henkyq) 发表:根据多年检测经验,ICP测土壤Cd有非常大的局限性,Cd的几个常用谱线(214 226 228)都会受到Fe元素的光谱干扰,会有叠加峰造成Cd检测结果虚高,而土壤里的Fe含量非常高(5%-15%),称样0.5g定容至50ml,Fe元素的含量一般在1000mg/L左右,会对Cd 214 产生强烈的光谱叠加,而光谱叠加是不能用标准加入法去除的,只能用干扰系数k值来抵消,但是因为每个样品土壤里的Fe含量并不十分接近,而Cd含量又太低,干扰系数有时候也无能为力,系统误差会大于实际值。 因此目前石墨炉测土壤Cd暂时还不能被ICP取代。 楼主测试的土壤Cd数据绝大部分并不是土壤的实际Cd值,而是Fe元素带来的干扰, 如果不信的话可以去检测一下这些样品的Fe含量,Fe含量高的Cd检测值就虚高。 需要验证方法可以采用国家标准土样GSS 或者ESS系列来检测,加标回收率并不适用于土壤这种复杂基体有光谱叠加可能性的样品。
后记:
由于这是我第一次进行土壤中重金属检测,没有相关数据参考,也没有相关经验可循,其中还有很多的不足,如:
1.加标回收率的处理,加标量应该与待测物的浓度水平相近,不得超过试样含量的3倍,一般是0.5-2倍,且不能超过线性范围的90%。而我则直接以二级标准作为加标量了。所以我在想,Pb,Cr的回收率好,Cd的回收率差是不是就是应为这个原因。还是说我加入的单标Cd失效,这个还需进一步验证。不知各位有什么见解?
2.标准曲线的设置问题,虽说ICP具有很宽的线性,且这次做下来,线性相关系数也很不错,但是针对具体样品,是不是还是应该适当的调整标系范围,特别是这次的Cd的标系,样品含量基本在曲线的1-5ug/L之间波动。
3.因为时间比较紧,这次测定完成后,土壤标准样品才姗姗来迟。前期准备不足。
这些问题,我将在后续的实验中逐一改正,也希望各位能多多指教,找出更多的不足之处,帮助我进步,先谢过了
原文由 zhaomin199(zhaomin199) 发表:学习了
根据多年检测经验,ICP测土壤Cd有非常大的局限性,Cd的几个常用谱线(214 226 228)都会受到Fe元素的光谱干扰,会有叠加峰造成Cd检测结果虚高,而土壤里的Fe含量非常高(5%-15%),称样0.5g定容至50ml,Fe元素的含量一般在1000mg/L左右,会对Cd 214 产生强烈的光谱叠加,而光谱叠加是不能用标准加入法去除的,只能用干扰系数k值来抵消,但是因为每个样品土壤里的Fe含量并不十分接近,而Cd含量又太低,干扰系数有时候也无能为力,系统误差会大于实际值。 因此目前石墨炉测土壤Cd暂时还不能被ICP取代。 楼主测试的土壤Cd数据绝大部分并不是土壤的实际Cd值,而是Fe元素带来的干扰, 如果不信的话可以去检测一下这些样品的Fe含量,Fe含量高的Cd检测值就虚高。 需要验证方法可以采用国家标准土样GSS 或者ESS系列来检测,加标回收率并不适用于土壤这种复杂基体有光谱叠加可能性的样品。