我做的相关实验美国药典已有收录(这句话好耳熟啊=。=),他用了标准加入法,一共4个点,A(定量限),B,C,D,分别加入0.5微克,1微克,2微克,0(未加入),也加了一定量的内标,纵坐标是峰面积比。A,B,C都能测到,D是未加入的,可能测不到,因为是杂质检查,这个杂质的量相当小,由A,B,C三点做一直线,外推此直线,延长线与X轴的交点与原点的距离即为药品中此杂质的含量。
以下是曾经泽老师(生物药物分析)中对标准加入法的描述
![]()
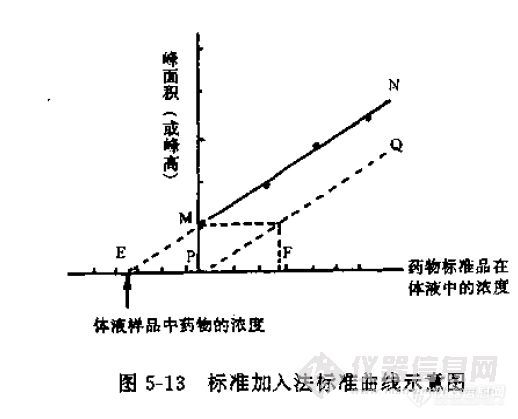
和示意图
![]()
与美国药典所阐述的一致。
我的问题是:1 如何证明A-C的斜率与小于A的某一浓度范围内的斜率是一样的? 换句话说由A,B,C三点做出的直线的斜率是K1,在小于浓度A(定量限)的某一范围内是测不出杂质的,它也有一个线,此线的斜率是K2,以上面的方法来看已经默认了K1=K2,那有没有直接的证据说明K1=K2?为什么要认为定量限以下的浓度所做得线性和之上的就一样呢?有没有相关书籍详细介绍了标准加入法的原理或方法的?
2 标准加入法的方法学怎么做?线性翻多少倍,加样回收率加多少样?怎么做?请详细说明
补充一句:美国药典之所以用标准添加去外推出此杂质的含量是因为此杂质含量很少且毒性也较大,限量是0.5ppm。我们只能比他低或一样,不能高
3 我还有个问题就是虽然做的是相同的样品,但由于仪器的不同导致定量限有很大的差别,人家是0.5微克/毫升,我们是15微克/毫升,比人家的高好多,如果以后只能用这种仪器来做,那我如何知道我的线性和人家低浓度的线性是否一致?更何况用低浓度的样品测得的数据本身就不稳定,稍有偏差,所得截距就会变化很大,所以我觉得还是从理论的源头研究,既然人家用了标准外加法,那我就得找到这种方法的依据,它在做线性的时候有没有什么规定翻多少倍?方法学怎么设计?等等