获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往
原文由 nini2006(nini2006) 发表:
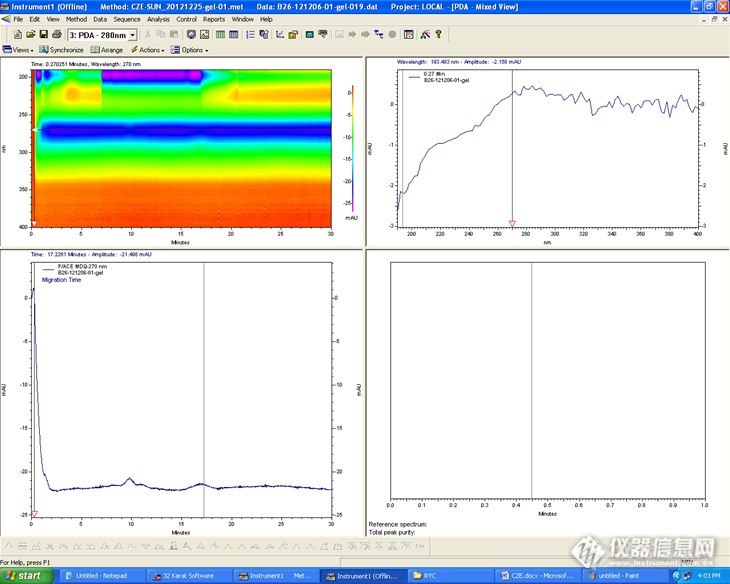
电流太小了,特别是硼酸的,提高浓度试试。可以参考下文献,看人家用的什么内径的管子还有用多大的浓度。
另外,当buffer的pH=pI的时候,蛋白质是不带电的,这样的话即使出峰也有可能跟其他中性物质混在一起分不开。
个人很不喜欢SDS,这个东西会把迁移时间拖长峰变宽,而且不容易控制重现性。当然,你既然用了,条件就要摸索才行。有时候只是因为你跑的时间不够,峰还没出来就被你停掉了。
原文由 threetigers107(threetigers107) 发表:既然有文献怎么会不知道时间?难道文献里居然一个图都没有?不同内径的管子对应不同的浓度,实在不行自己摸索。酶就是蛋白质的一种,可以借鉴蛋白质的分离模式。蛋白质的分离用等电聚焦电泳的比较多,当然也有用SDS的胶束电动色谱,但是如果用的silica-fused capillary吸附比较厉害,可以考虑用其他无胶筛分模式和动态涂敷的方式。原文由 nini2006(nini2006) 发表:
电流太小了,特别是硼酸的,提高浓度试试。可以参考下文献,看人家用的什么内径的管子还有用多大的浓度。
另外,当buffer的pH=pI的时候,蛋白质是不带电的,这样的话即使出峰也有可能跟其他中性物质混在一起分不开。
个人很不喜欢SDS,这个东西会把迁移时间拖长峰变宽,而且不容易控制重现性。当然,你既然用了,条件就要摸索才行。有时候只是因为你跑的时间不够,峰还没出来就被你停掉了。
谢谢一楼帮我贴图。昨天搞了老半天不知道怎么弄上去,主要是电脑太慢了。。。
看了一些文献,一般buffer用的浓度也不是太高,20,50,100mM不等;毛细管主要有50和75两种粒径的,长度40,50cm不等;分离时间的话就不太清楚了。不知道各位一般是多长时间的。我做的是一个多聚体的酶和一个peg化的样品