获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往
原文由 小百度12597(v2736091) 发表:原文由 尘(fjh26) 发表:原文由 小百度12597(v2736091) 发表:原文由 尘(fjh26) 发表:
做汞,我的氢氧化钠是0.5%,硼氢化钾1%,在流盐酸5%,
一级气液分离器才要液封,为了防止生成的氢化物从废液管路跑走[/quote
你好,液封是跟气液分离器单独分开的一个装置是吗??我那除了气液分离器就没有别的装置了,对了您方便给我留个QQ吗?
液封不是一个装置,就是往气液分离器里加水
我这机子里有两个气液分离器,是往两个企业分离器里都加水吗??加水量多少为合适?
加前一个就行,原子化器下面的那个不能有液体的
原文由 尘(fjh26) 发表:原文由 小百度12597(v2736091) 发表:原文由 尘(fjh26) 发表:原文由 小百度12597(v2736091) 发表:原文由 尘(fjh26) 发表:
做汞,我的氢氧化钠是0.5%,硼氢化钾1%,在流盐酸5%,
一级气液分离器才要液封,为了防止生成的氢化物从废液管路跑走[/quote
你好,液封是跟气液分离器单独分开的一个装置是吗??我那除了气液分离器就没有别的装置了,对了您方便给我留个QQ吗?
液封不是一个装置,就是往气液分离器里加水
我这机子里有两个气液分离器,是往两个企业分离器里都加水吗??加水量多少为合适?
加前一个就行,原子化器下面的那个不能有液体的
\前一个是一级气液分离器,要是往里加水的话,它不会从下面的管子排废液排出去吗??对了能不能请教一个问题,如果标准溶液用硝酸定容的话,用多大浓度的合适?如果用盐酸定容的话,浓度是跟载流浓度一样吗,就是5%??然后里面还用加其他东西吗??
原文由 小百度12597(v2736091) 发表:原文由 尘(fjh26) 发表:原文由 小百度12597(v2736091) 发表:原文由 尘(fjh26) 发表:原文由 小百度12597(v2736091) 发表:原文由 尘(fjh26) 发表:
做汞,我的氢氧化钠是0.5%,硼氢化钾1%,在流盐酸5%,
一级气液分离器才要液封,为了防止生成的氢化物从废液管路跑走[/quote
你好,液封是跟气液分离器单独分开的一个装置是吗??我那除了气液分离器就没有别的装置了,对了您方便给我留个QQ吗?
液封不是一个装置,就是往气液分离器里加水
我这机子里有两个气液分离器,是往两个企业分离器里都加水吗??加水量多少为合适?
加前一个就行,原子化器下面的那个不能有液体的
\前一个是一级气液分离器,要是往里加水的话,它不会从下面的管子排废液排出去吗??对了能不能请教一个问题,如果标准溶液用硝酸定容的话,用多大浓度的合适?如果用盐酸定容的话,浓度是跟载流浓度一样吗,就是5%??然后里面还用加其他东西吗??
会排出去但不会全部出去,都会有一定量的残留的,所以不管你加多少量都一样的,除非管线方向连错全部排出去了
标准溶液尽量和载流一样,汞的话不用加,其他如砷、锡等都要加还原剂的,具体参考标准
原文由 wangjunyu(wangjunyu1113) 发表:
你们的仪器是什么时候买的啊,他们家新的仪器是不用水封的。原子荧光的空白值在那上下波动也是正常的,特别是测汞。你测量砷的标准曲线好不好呢,
汞的标准曲线你不一定要天天配置的,一定要注意在配置过程中所有的器皿都要浸泡干净。标准曲线测好了,测量控制样才能准确。
原文由 一土(asoil) 发表:
有信号,没漏气。线性可以,稳定性可以。标准空白六百多,有点高,看是否有污染或水试剂纯度。1,荧光值五百多,有点低,优化进样反应系统,荧光系统,看峰形读数延迟是否合理。
原文由 小百度12597(v2736091) 发表:
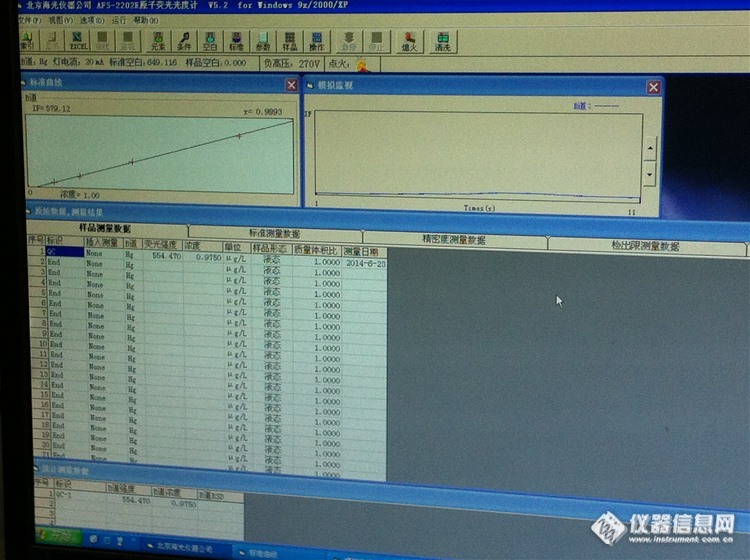
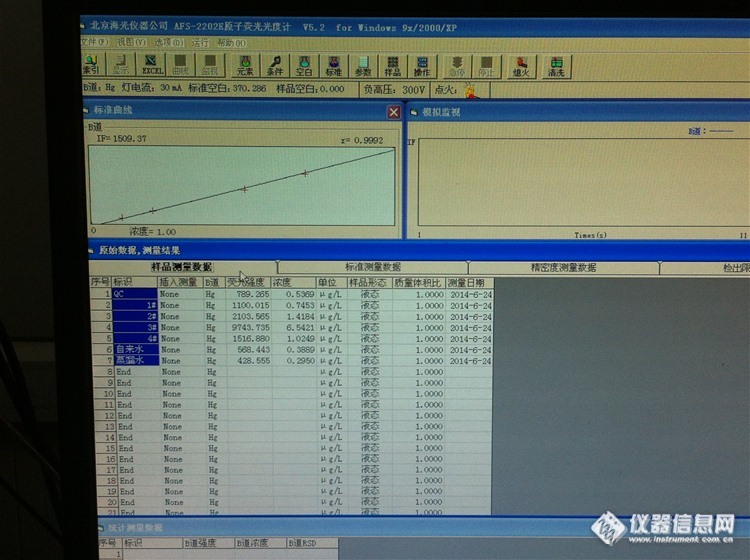
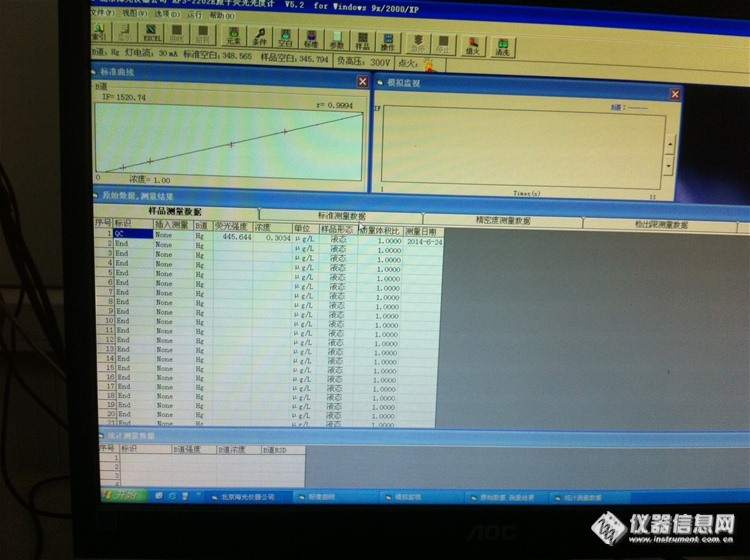
你好,怎么样看峰形看读数延迟是否合理??他俩又什么联系??后来我又测了下,空白下来了,三四百左右,线性也可以,但是我没测样品空白,直接测得质控样,浓度是0.5,标准浓度就是0.5. 但是后来人家说不能不测样品空白,我又测得样品空白,在走的质控样,浓度是0.3. 低了好多,从新走了好几次都是这样,请问什么情况,哪里出了问题?
原文由 小百度12597(v2736091) 发表:
你好,怎么样看峰形看读数延迟是否合理??他俩又什么联系??后来我又测了下,空白下来了,三四百左右,线性也可以,但是我没测样品空白,直接测得质控样,浓度是0.5,标准浓度就是0.5. 但是后来人家说不能不测样品空白,我又测得样品空白,在走的质控样,浓度是0.3. 低了好多,从新走了好几次都是这样,请问什么情况,哪里出了问题?