维权声明:本文为Insm_df8bf4f4原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。
Part1:理论部分衍生方法使被测物与相应的试剂发生化学反应从而改变被测物的理化性质。
(一)HPLC衍生化目的
①改善被测物的检测
②改变被测物的分子结构或极性,以利于色谱分析
③改变基质,以利于色谱分析
④改善被测物的不稳定性
(二)衍生试剂选用原则
①衍生试剂必须稳定
②衍生化中,衍生产生的副产物不干扰目标物的
分离和检测
③被测物与衍生试剂发生反应的条件不能太苛刻
④衍生试剂尽量无毒或低毒
⑤步骤尽量适于自动化,
容易操作(三)衍生方法
根据衍生反应的场所来分,可分为柱前衍生和柱后衍生。
A、柱前衍生(pre-columnderivatization)①紫外衍生化表1:能提高UV检测的有关发色团| 发色团 | 最大吸收波长(nm) | 254nm处摩尔吸收系数 |
| 苯甲基(苄基) | 254 | 200 |
| 4-硝基苄基 | 265 | 620 |
| 3,5-二硝基苄基 | - | >10000 |
| 苯甲酸酯(或盐) | 230 | 低 |
| 4-氯苯甲酸酯 | 236 | 6300 |
| 4-硝基甲酸酯 | 254 | >10000 |
| 2,4-二硝基苄基 | - | >10000 |
| 甲苯酰基 | 236 | 5400 |
| 甲氧苯(苄)基 | 262 | 16000 |
| 苯甲酰甲基 | 250 | 10000 |
| 4-溴苯甲酰甲基 | 260 | 18000 |
| 2-萘 | 248 | 16000 |
表2:常用UV衍生剂| 名称 | 最大吸收波长(nm) | 摩尔吸收系数 |
| 2,4-二硝基氟苯 | 350 | >104 |
| 对硝基苯甲酰氯 | 254 | >104 |
| 对甲基苯磺酰氯 | 224 | 104 |
| 异硫氰酸苯酯 | 244 | 104 |
| 对硝基苯基溴 | 265 | 6200 |
| 对溴代苯甲酰甲基溴 | 260 | 18000 |
| 萘酰甲基溴 | 248 | 18000 |
| N,N对硝基苄基异丙基异脲 | 265 | 6200 |
| 3,5二硝基苯甲酰氯 | 248 | 104 |
| 对甲氧基苯甲酰氯 | 262 | 16000 |
| 2,4二硝基苯肼 | 254 | 18000 |
| 对硝基苯甲氧胺盐酸盐 | 254 | 6200 |
表3:常用的紫外衍生反应| 反应类型 | 反应产物 |
| 苯甲酰化反应 | 对硝基苯甲酰氯、3,5-二硝基苯甲酰氯和对甲氧基苯甲酰氯与胺、醇、酚类化合物反应,生成强紫外吸收的苯甲酸酯类衍生物 |
| 2,4-二硝基氟代苯反应 | 与大多数伯胺、仲胺和氨基酸反应,生成强紫外吸收的苯胺类衍生物 |
| 苯基异硫氰酸酯的反应 | 与氨基酸、醇类反应,生产对应的苯基硫酸盐 |
| 苯基磺酰氯的反应 | 与伯胺、仲胺反应,生产苯基磺酰胺 |
| 有机酸的酯类反应 | 有机酸与酰溴基试剂反应成酯 |
| 羰羟基化合物的反应 | 醛类、酮类的羰基与2,4-二硝基苯肼反应,生成苯腙衍生物;与对硝基苄基羟胺反应。 |
②荧光衍生化在
液相色谱中,荧光检测器是一种高灵敏度、高选择性的检测器,比紫外检测器的灵敏度要高10-1000倍。因此,可以检测痕量非荧光物质,常将它与荧光衍生化试剂反应,生成荧光物质,用荧光检测器检测。
表4:常用荧光衍生化试剂| 名称 | 激发波长(nm) | 发射波长(nm) |
| 丹磺酰氯 | 340 | 355 |
| 丹磺酰肼 | 340 | 525 |
| 荧光胺 | 340 | 525 |
| 邻苯二甲醛 | 340 | 455 |
| 4-溴甲基-7-甲氧基香豆素 | 365 | 420 |
| 芴代甲氧基酰氯 | 260 | 310 |
| 荧光素异硫氰酸酯 | 350 | 383 |
| 4-氯-7-硝基苯一氧二氮杂茂 | 380 | 530 |
PS:衍生化的荧光波长范围与荧光试剂和副产物不同,荧光衍生反应不需纯化衍生物,可以直接进样③手性衍生化用手性试剂与外消旋体反应,在分子内导入另一手性中心,柱前衍生反应生成一对非对映异构体,两者间无镜像关系,物理化学性质不同,可用常规的色谱分离条件进行分离。
表5手性衍生应用范围| 条件 | 衍生方法 |
| 不宜直接拆分 | 如游离胺类在手性固定相上往往呈较弱的色谱性质,生成中性化合物则获得显著改善; |
| 对映异构选择性差 | 添加某些基团,可增加色谱系统的对映异构选择性 |
| 检测器响应差 | 提高紫外或荧光检测的效果 |
表6:手性分析的官能团及衍生物| 官能团 | 衍生试剂 |
| 氨基 | 胺、氨基甲酸酯、脲、硫脲、硫酰胺 |
| 羟基 | 酯、羧酸、碳酸酯、氨基甲酸酯 |
| 羧基 | 酯、胺 |
| 环氧化物 | 异硫氰酸酯、烯烃(手性铂络合物) |
| 硫醇 | 硫酯 |
B、柱后衍生(post-columnderivatization)样品先进入色谱柱,经分离,各个组分从色谱柱流出后,与衍生化试剂反应,在一定反应条件下,生成具有某种检测特性的产物,再进入检测器,得到检测信号。
①柱后衍生法的优点a.形成副产物不重要,反应不需要完全,产物也不需要高的稳定性,只需要有好的重复性即可;b.被分析物可以在其原有的形式下进行分离,容易选用已有的分析方法。②柱后衍生法的缺点a.对于一定的溶剂和有限的反应时间来说,目前只有有限的反应可供选择;b.需要额外的设备,反应器可造成峰展宽,降低分辨率。③柱后衍生设备a.毛细管式柱后反应器
b.空气分割式柱后反应器
c.填充管式反应器
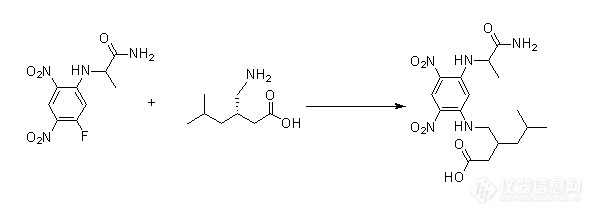
Part2:案例分享(一)案例1(手性异构分析)衍生理由:此氨基酸紫外吸收很弱,直接紫外检测灵敏度低;
此氨基酸为对映异构体,普通的反相色谱条件无法分离。
①衍生原理![]() ②衍生方法分别移取1mL供试品溶液(20mg供试品加水稀释至10mL)、1mL衍生化化试剂(50mgNα-(5-氟-2,4-二硝基苯基)-L-丙氨酰胺加乙腈稀释至10mL)到10mL容量瓶,混匀后再加入100μLNaHCO3(0.84gNaHCO3加水稀释至10mL)溶液,盖上盖子,置于40℃,搅拌模式下衍生反应1小时后加入100μLHCl溶液终止反应;取其中200μL反应液加800μL流动相,混匀过滤后作为供试品测试溶液。③色谱条件
②衍生方法分别移取1mL供试品溶液(20mg供试品加水稀释至10mL)、1mL衍生化化试剂(50mgNα-(5-氟-2,4-二硝基苯基)-L-丙氨酰胺加乙腈稀释至10mL)到10mL容量瓶,混匀后再加入100μLNaHCO3(0.84gNaHCO3加水稀释至10mL)溶液,盖上盖子,置于40℃,搅拌模式下衍生反应1小时后加入100μLHCl溶液终止反应;取其中200μL反应液加800μL流动相,混匀过滤后作为供试品测试溶液。③色谱条件色谱柱:Hypersil BDS C18,4.6mm×150mm×5um
流动相A: 1%三乙胺磷酸调pH到3.0流动相B:乙腈
进样体积:20ul
柱温:30℃,流速:2.0ml/min,
检测波长:340nm
流动相A:流动相B=62:38
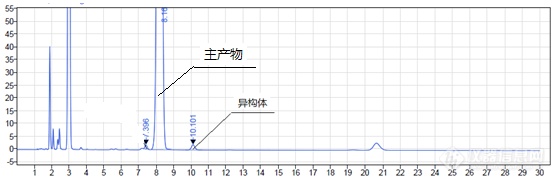
运行时间:25min④典型图谱![]() (二)案例2(盲样衍生方法选择)
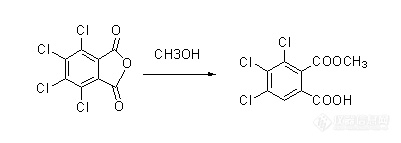
(二)案例2(盲样衍生方法选择)此结构(酸酐)在
液相中不稳定,遇水变成羧酸,而本反应目标产物就是羧酸,因而需要衍生;
①衍生条件初选与确定a.起初用吗啡啉衍生,衍生产物和产品分离度不佳,并且吗啡啉衍生很困难(吗啡啉位阻较大)最终选用甲醇衍生。
b.考虑到样品与甲醇溶解性不好的问题,样品用甲醇衍生的时候配成了30%的甲醇乙腈溶液。
衍生反应方程式:
![]() ②衍生条件确定
②衍生条件确定a.先找时间最佳值(温度固定位60℃),10~60min(10min间隔), 实验数据显示最佳时间为30分钟 ;
b.找最佳温度(时间固定位30分钟) 60℃、70℃、80℃,实验数据显示最佳温度为70℃;
c.确定最终衍生条件:70℃下衍生30分钟为最佳组合。
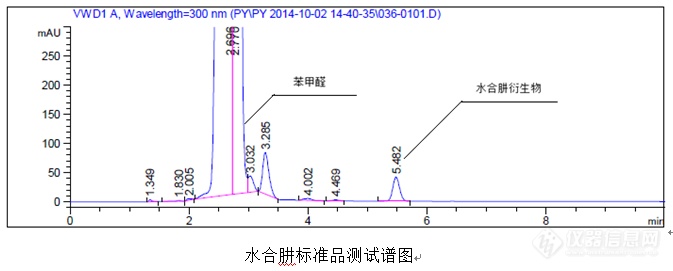
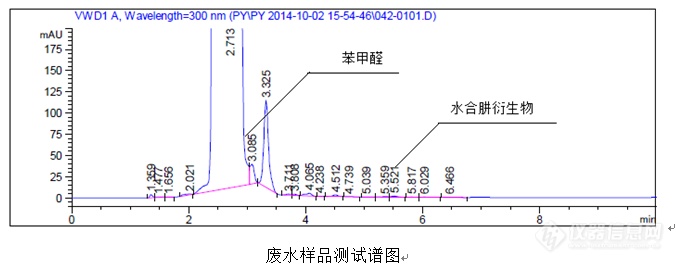
(三)案例3(衍生外标法运用)案例:HPLC法测定废水中水合肼含量①试剂试药甲醇(HPLC级)乙腈(HPLC级)四氢呋喃(分析纯)苯甲醛乙二胺四乙酸二钠(EDTA)纯化水80%的水合肼水溶液(需进行滴定测定准确含量后使用)②色谱条件色谱柱:Agilent HypersilODS,250mm×4.0mm×5.0μm 流动相A:300mg乙二胺四乙酸二钠,700ml乙腈,300ml水洗脱程序为:等度检测波长:300nm流速:1.0ml/min柱温:30℃运行时间:10min③衍生操作过程储备液1:准确称取125.00mg(精确至0.01mg)水合肼标准品至100mL容量瓶,加入乙腈稀释至刻度,摇匀备用。储备液2:准确移取1mL储备液1至100mL容量瓶,乙腈稀释至刻度,摇匀备用。储备液3:准确移取1mL储备液2至10mL容量瓶,乙腈稀释至刻度,摇匀备用。标准品溶液:准确移取1mL储备液3至10mL容量瓶,移液枪准确移取150μL苯甲醛至其中,加入1.5mL四氢呋喃,3mL甲醇,1mL乙腈,0.5mL水,超声10min,待冷却后甲醇稀释至刻度,摇匀备用。(注意:一定要按照上述顺序依次加入溶剂)样品溶液:准确称取5000.00mg(精确至0.01mg)样品至10mL容量瓶,移液枪准确移取150μL苯甲醛至其中,加入1.5mL四氢呋喃,3mL甲醇,1mL乙腈,0.5mL水,超声10min,待冷却后甲醇稀释至刻度,摇匀备用。(注意:一定要按照上述顺序依次加入溶剂,否则样品不溶解)空白溶液:乙腈:水=7:3④水合肼(wt%)计算公式![]() ⑤典型图谱
⑤典型图谱![]()
![]()
申明:本文为个人经验分享仅代表个人观点,仅供参考,欢迎交流!
更多精彩内容,请关注微信公众号“研发分析之路”