获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往

原文由 tuxlin(tuxlin) 发表:欢迎常来参与讨论
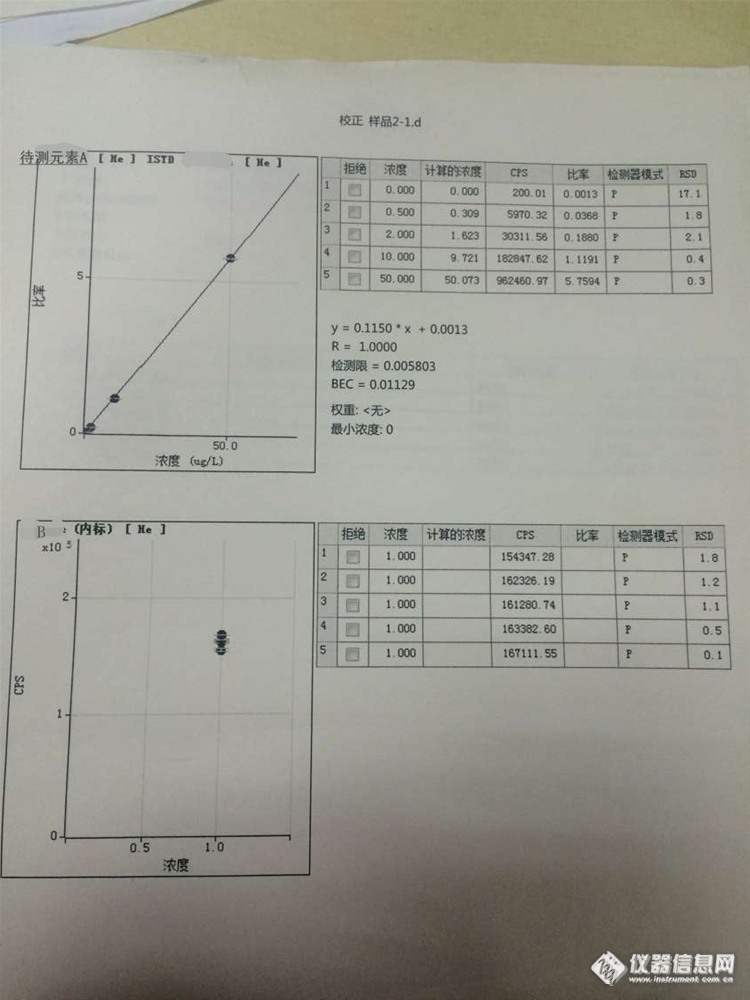
1、计算过程就是你上面说的那样。你的这个试样空白太高了,在样品含量的一半左右。
2、内标在含量的计算过程中是有作用的。观察标准曲线就会发现,加了内标的话。纵轴是比率(元素计数/内标计数),计算样品的含量也是用样品计数/内标计数的比率。而不加内标的话,纵轴就是元素计数,计算含量就直接用样品的计数。
理论上来讲,内标的浓度就与待测元素的浓度相近。但实际中待测样品中的含量千差万别,是很难兼顾到的。A的工程师是建议用比较高的内标浓度。个人认为控制在十几万-三十万CPS左右较好。
3、结果与厂家报告给的差倍数的话,肯定是计算的问题,再检查一下计算过程吧。
原文由 tuxlin(tuxlin) 发表:
1、计算过程就是你上面说的那样。你的这个试样空白太高了,在样品含量的一半左右。
2、内标在含量的计算过程中是有作用的。观察标准曲线就会发现,加了内标的话。纵轴是比率(元素计数/内标计数),计算样品的含量也是用样品计数/内标计数的比率。而不加内标的话,纵轴就是元素计数,计算含量就直接用样品的计数。
理论上来讲,内标的浓度就与待测元素的浓度相近。但实际中待测样品中的含量千差万别,是很难兼顾到的。A的工程师是建议用比较高的内标浓度。个人认为控制在十几万-三十万CPS左右较好。
3、结果与厂家报告给的差倍数的话,肯定是计算的问题,再检查一下计算过程吧。