获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往

原文由 anping 发表:
塞曼效应原子吸收技术的原理(摘自生命经纬网)
塞曼效应原子吸收技术即利用塞曼效应作背景校正进行双光束测量。
当具有适当强度的磁场作用于原子化器所产生的原子蒸汽时,主吸收谱线因塞曼效应而被分裂成三种成分:π成分(△M=0)和σ±成分(△M=±1)。其中π成分和σ±成分是分别与磁场平行或垂直的偏振光束。在正常的塞曼效应中,π谱线的波长无变化,因而只有 π谱线与空心阴极灯的发射谱线相匹配,σ±谱线则偏离发射谱线。如此即有:
(1)偏振的共振发射线中的P∥成分被原子蒸汽的π谱线所吸收。
(2)由于σ±谱线漂移对共振发射线中的P⊥成分的原子吸收灵敏度降低。
由分子吸收和光散射引起的背景吸收不受塞曼效应和影响,因而P∥和P⊥成分均被背景等量吸收。原子吸收加上背景吸收用P∥成分测量,而微弱的原子吸收加上背景吸收用P⊥成分测量,求出上述测量值之差,就可获得原子吸收的测量值。
责任编辑:合欢巢蛾 BIOX.CN 2006-8-24 22:40 来源:生命经纬网
原文由 jackdoason 发表:
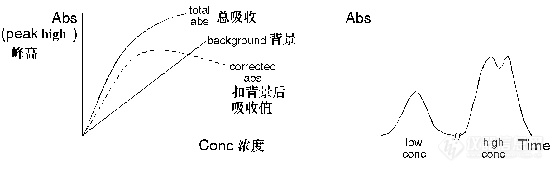
在较高浓度时,校正曲线通常是趋于某一极限值。但在塞曼系统中,校正曲线(采用峰 高法)可能出现向下翻转的情况(依据波长不同弯曲程度不一),这样就会有两个浓度 值对应同一个吸光度值的现象发生?
请问此种情况如何解决?
原文由 handsomeland 发表:原文由 shaweinan 发表:原文由 anping 发表:
样品光束(信号)的振动方向是平行磁场的;背景光束(信号)的振动方向是垂直磁场的;也就是说,两束光虽然是合并为一条(暂且这样形容)光束,但关键是振动方向不一致,两者相差90度。分束镜外形类似三棱镜,但它的两个斜面却类似光栅,表面(底面?)有刻槽,刻槽的方向与入射的光束方向有共振关系(这是我的理解,我没有这方面的资料),于是分束镜利用共振关系,就会将两束光分裂开,并由各自的检测器对应接收。
需要解释的是,关于分束镜的原理和构造的资料我没有,请网友及shaweinan先生见谅!
我还是真没有看清分束镜是个三棱镜,只看到它的底座了。所以从光路上看它应该是出来的光束是正对在三棱镜的中间的那条边上,将光一分为二。不知有没有这个分束镜更清晰一些的照片,我个人认为它应该没有检偏作用,它的两个斜面上真的有刻槽吗?如果是的话那应该对光产生的是衍射作用呀,还能起到检偏作用?我对检偏器的原理不是十分了解,不过印象中好象是利用某些具有旋光性物质的特性来对光进行检偏的,而且是透过检偏,所以我认为检偏器很可能在检测器的前面。从这种光路结构来看,那它同样是以减小一半光能量为代价的。这个仪器应该采用的是恒定磁场,如果磁场是加在原子化器的地方,那它不象变化磁场方式,在无磁场时测量元素+背景吸收有磁场时测背景吸收,灵敏度损失比较大。再有就是这种光路设计,因为出射光要正好对在三棱镜的中间那条边上,所以如果不是卡式结构,那调整校正起来就不是很容易。
根据我的理解,分光镜只是一只普通的三棱镜,底面是镜面,斜面透明,都没有刻槽,即从分光镜出来的两束光偏振特性还是一样的,偏振的区分恐怕在探测器的前端。能够对多波长的偏振进行分离的三棱分光镜我还没见过。
原文由 camel1998 发表:
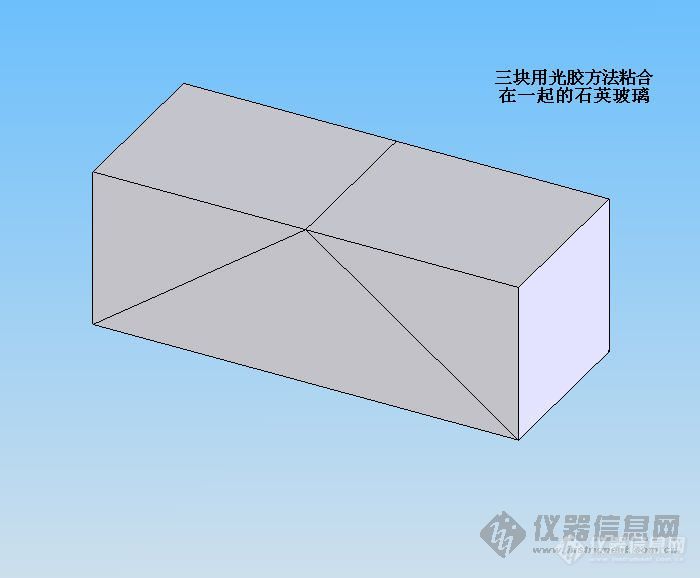
我来简单解释一下shaweinan的疑问,直流塞曼原子吸收的调制方法是偏振调制。所谓偏振调制,就是在光束出单色器的时候以特定角度经过一块沃拉斯顿棱镜(棱镜的行装如图所示),经过棱镜后,两个不同偏振方向的光会因为偏振方向和沃拉斯顿棱镜的石英玻璃的晶轴方向重合或垂直而发生折射角不同的折射,从而被分开一个很小的角度。这个角度虽然很小,但是随着光程的增加,两种偏振成分光斑的距离会增大,知道两个光斑能被完全分开,经过一个反射棱镜反射后,分别入射到两个检测其中。不知我这样解释shaweinan和其他网友能不能明白。