后续将有专属客服与您沟通!
关注微信公众号查看留言进度 接收留言处理通知
0
ID:xx_dxd_xx
行业:其他
积分:0升级还需100积分
声望:0升级还需100声望
注册时间:0000-00-00
最后登录时间:0000-00-00
请确认联系方式
请输入您的联系方式
提交留言即视为您同意遵守 《服务协议》和 《隐私权政策》
ID:jimzhu
ID:v3237657
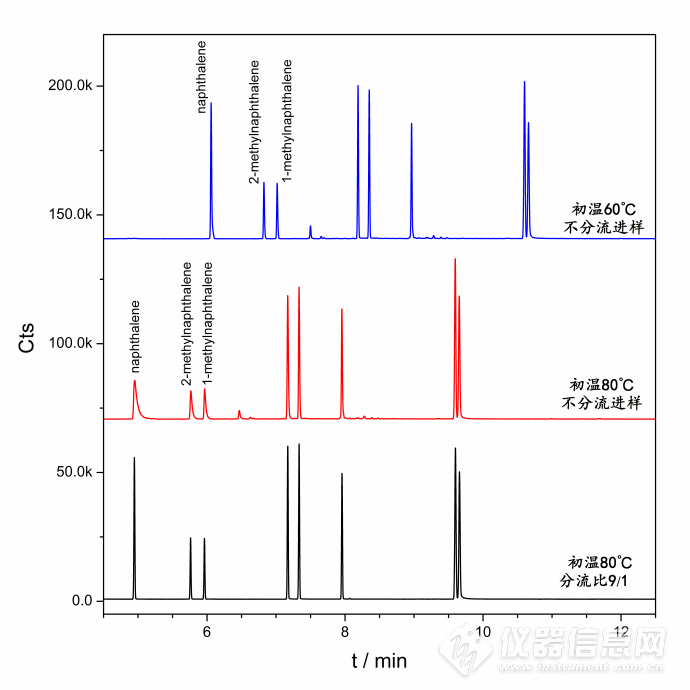
原文由 xx_dxd_xx(xx_dxd_xx) 发表:色谱条件好像写少了一行,不知道还能不能修改
ID:colray
原文由 symmacros(jimzhu) 发表:以前做多环芳烃不仅仅离子源温度调高,四极杆温度也调高了一点。
ID:v3333209
原文由 xx_dxd_xx(xx_dxd_xx) 发表: 提高四级杆温度是不是为了防止污染?我这个是5973的老仪器,四级杆好像150就是上限了