维权声明:本文为henkyq原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。
前言:
不知道各位有没有这样的体验,明明某些元素的测定条件已经优化调试的好好的,满心以为可以应对日常的检测工作了。但是隔几天就会冒出一些样品向你的优化发出挑战,让你知道不是所有的样品都可以用一个测定条件就能打发的。前一段时间一组盲样中铅元素测定的考核就让这故事重演了。
前处理:
消化管中先加2ml硝酸,再取样1.00g,再补齐硝酸至10ml,加三颗玻璃珠,盖好塞子放入石墨消解炉中,过夜冷消化,再程序升温消解,80℃60min,150℃60min,打开塞子,190℃90min消化至近干,用1%HNO3溶液定容至25ml待测。大葱标样和一组盲样消化液浑浊,肉眼可见有小颗粒物,另一组盲样消化液则清亮透明。所有消化液静置沉淀后取上清液上机测定。
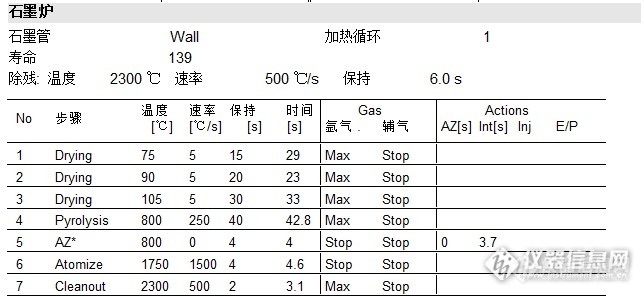
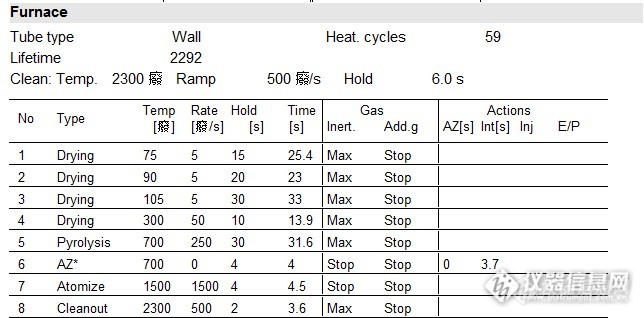
按照平时的食品测定方法检测(升温程序见下图1),发现线性满足要求,但是标样及盲样均无法测出,甚至比曲线空白还低。考虑是不是消化不彻底的原因,但是石墨炉不像火焰,对样品的消化彻底程度要求没那么高,毕竟石墨炉有一个程序升温程序过程,在一定程度是也是起到前处理作用的。那就有可能是我的测定程序有欠缺。
逐步排查,首先利用原有消化液再次针对具体样品优化测定方法,其次再优化消化过程。查阅相关文献后,有一篇文章提到一组基体改进剂:2% NH4H2PO4+1%Mg(NO3)2,文献说明如下:NH4H2PO4能增加铅的稳定性,提高灰化温度,是基体中难挥发的盐转化为易挥发的盐,使其在原子化之前更多的蒸发溢出石墨管,从而使基体效应的干扰降低。Mg(NO3)2是一种良好的助灰化剂,在灰化过程中能产生NO2和MgO,前者能促进样品溶液中有机物氧化分解,减小原子化时的背景干扰,后者壳稀释灰分,使灰分均匀分布,有利于提高测定的重现性。考虑到我平时都是用1% NH4H2PO4做为基改剂,这次就试试文中提到的基改剂组合2% NH4H2PO4+1%Mg(NO3)2,顺便对比两组基改剂。
![]()
图1
![]()
图2
![]()
图3
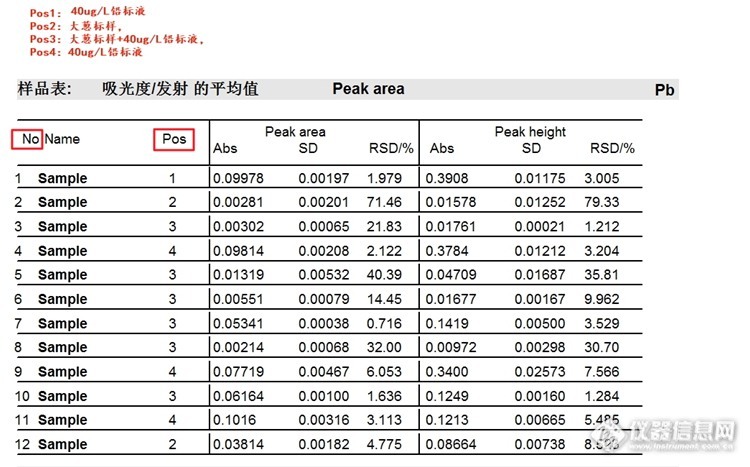
在图2中:(
No为样品的测定顺序,Pos为样品杯所在的位置,注意区分哦!)
No1-No4:1% NH
4H
2PO
4,为平时的食品检测升温程序,见图1
No5:1% NH
4H
2PO
4,增加干燥步骤+300℃10s,灰化800℃不变,时间改为30s
No6:1% NH
4H
2PO
4,增加干燥步骤+300℃10s,灰化改为900℃,时间改为30s
No7:2% NH
4H
2PO
4+1%Mg(NO
3)
2,增加干燥步骤+300℃10s,灰化800℃时间30s
No8:2% NH
4H
2PO
4+1%Mg(NO
3)
2,增加干燥步骤+300℃10s,灰化900℃时间30s
No9:2% NH
4H
2PO
4+1%Mg(NO
3)
2,增加干燥步骤+300℃10s,灰化900℃时间30s
No10-No12:2% NH
4H
2PO
4+1%Mg(NO
3)
2,增加干燥步骤+300℃10s,700℃灰化30s,1500℃原子化(优化后的升温程序,过程见下图4)
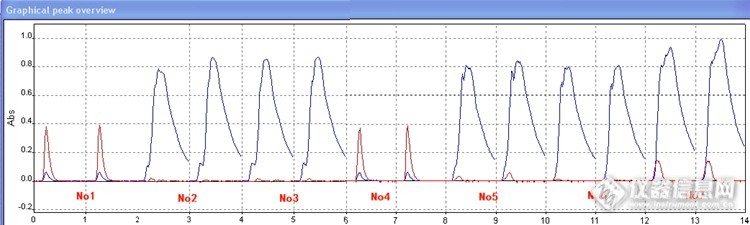
依据图2-图3做出数据分析:
1. 从样品No1-No4的实验数据可以得出采样平时的检测程序,样品含有基体时对实验测定有明显的干扰
2. 分析数据No5-No6以及No7-No8可以看出在基改剂一样,同时都增加一步干燥步骤300℃10s时,灰化温度800℃对应的吸光度比900℃对应的吸光度要高。
3. 对比数据No5和No7可以看出,在升温程序一样时,基改剂2% NH
4H
2PO
4+1%Mg(NO
3)
2对应的吸光度明显高于1% NH
4H
2PO
4对应的吸光度。
4. 分析整个数据可以看出No10是样品Pos3中吸光度最高的,No12是样品Pos2中吸光度最高的,而No11较以前的升温程序测出的吸光度差异不大。
小结:
更改基改剂以及优化升温程序后,对含有基体干扰的样品测定而言有明显的改善,但是对不含基体的标液而言变化不大。
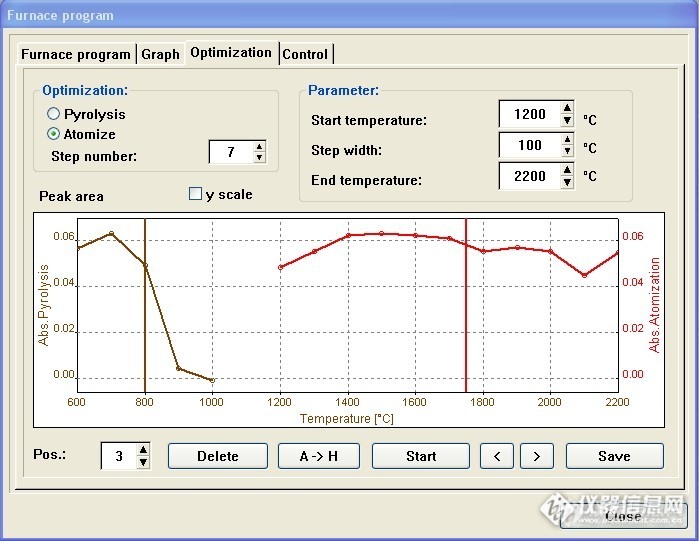
以Pos3(大葱标样+40ug/L铅标液)为样品,2% NH
4H
2PO
4+1%Mg(NO
3)
2为基改剂,进行升温程序优化,如图4:(说明:未勾选“y scale”选项,就未显示背景,同时未切换峰高视图)
![]()
图4
根据上图选择700℃灰化,1500℃原子化,完整升温程序见下图5:
![]()
图5
用以上优化后的方法,同时做标准曲线法和标准加入法
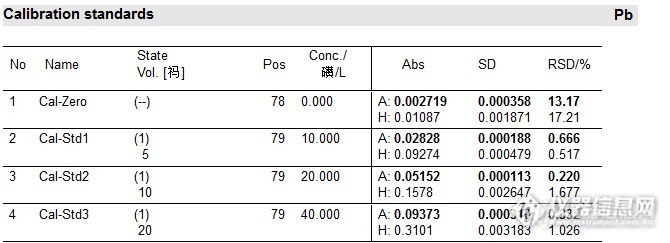
标准曲线数据如下图6-7:
![]()
图6
![]()
图7
测试数据如下图8 :
![]()
图8
No1-No3:分别为20ug/L铅标液、稀释2倍的大葱标样、稀释2倍的(大葱标液+40ug/L铅标液混合液)
No4:稀释2倍的大葱标样
No5:稀释2倍的大葱标样+20ug/L铅标液
No6:稀释2倍的大葱标样+40ug/L铅标液
No7:稀释2倍的大葱标样+80ug/L铅标液
即No4-No7为大葱标液的标准加入法测定过程。
从样品No1,2,4可以看出大葱标样的铅含量和40ug/L铅标液接近,但是按照以往的实验来看,铅含量高于40ug/L时会出现曲线弯曲,不成线性,那么这次的加标量也就过高,而且从外标曲线得出的铅含量来看也不是成比例增加的。
![]()
图9
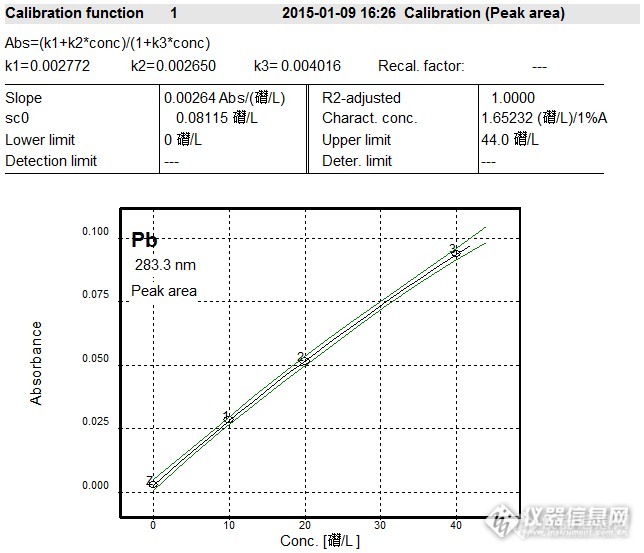
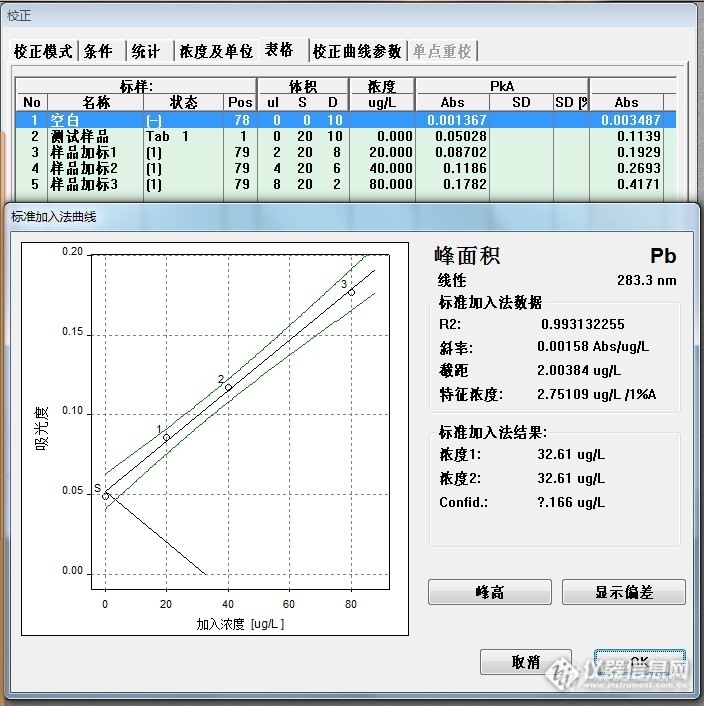
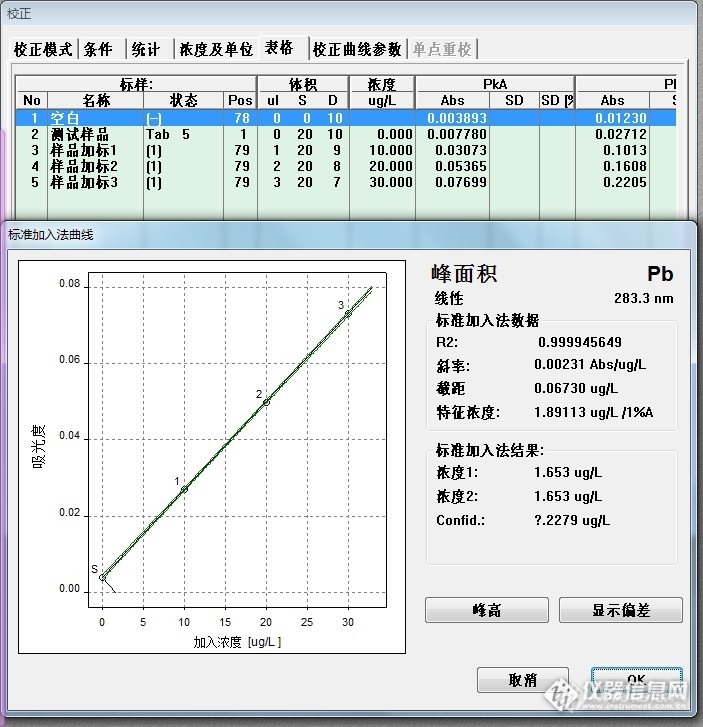
标准加入法线性回归:样品含量为32.61*2=65.22ug/L.(图9中的“空白”1%硝酸溶液未做,就按照以往的实验数据填写了一组,也未做样品消化空白,就无法减空白)。
大葱标样的铅参考值为1.34±0.16mg/kg ,理论计算:取样1.00g,消化定容至25ml,样品消化空白为0ug/L,那么大葱铅含量为53.6±6.4ug/L,该组测试数据超出参考范围。
重新调整加标浓度,同时补全数据,样品消化空白,1%HNO
3空白,数据如下图10:
![]()
图10
图10中的样品说明:
No1:样品消化空白
No2:样品消化空白+10ug/L铅标液
No3:样品消化空白+20ug/L铅标液
No4:样品消化空白+30ug/L铅标液
No5:稀释2倍的大葱标样
No6:稀释2倍的大葱标样+10ug/L铅标液
No7:稀释2倍的大葱标样+20ug/L铅标液
No8:稀释2倍的大葱标样+30ug/L铅标液
No9:1%HNO
3曲线回归时共用的空白
No10:20ug/L的铅标液
![]()
图11
样品消化空白回归结果:1.653ug/L
(线性非常好,说明样品中目标元素与加入的标准总含量在40ug/L以内曲线呈线性)
![]()
图12
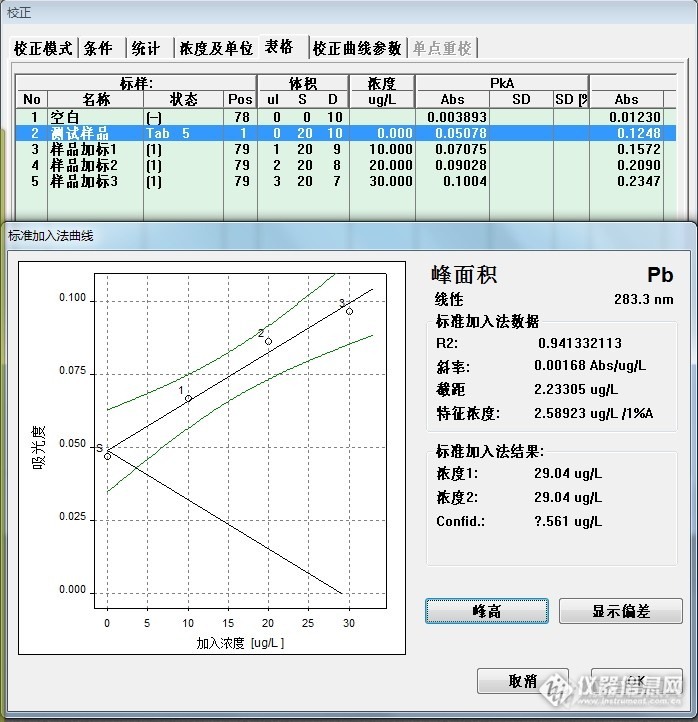
大葱标样回归结果:29.04ug/L*2=58.08ug/L
代入公式计算:X=(c-c
0)*v/(1000*m); v=25ml
大葱取样为1.00g,计算结果为:1.41mg/Kg,大葱标样的铅参考值为1.34±0.16mg/kg,符合要求。
(线性较差,最后一点样品中目标元素与加入的标准总含量在40ug/L以上,曲线出现弯曲,某种程度上也可以说明标准加入法也应该是呈线性使用。)
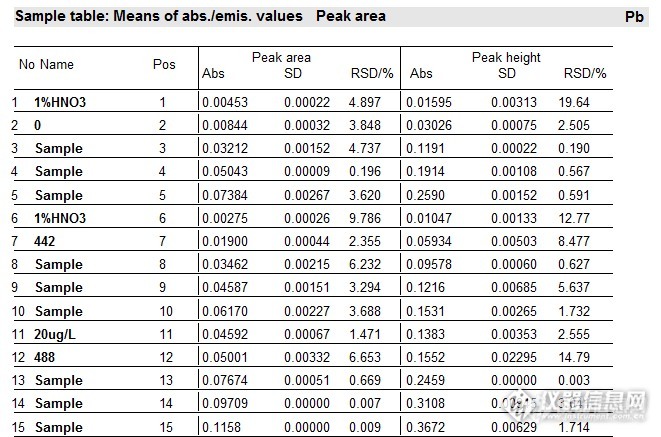
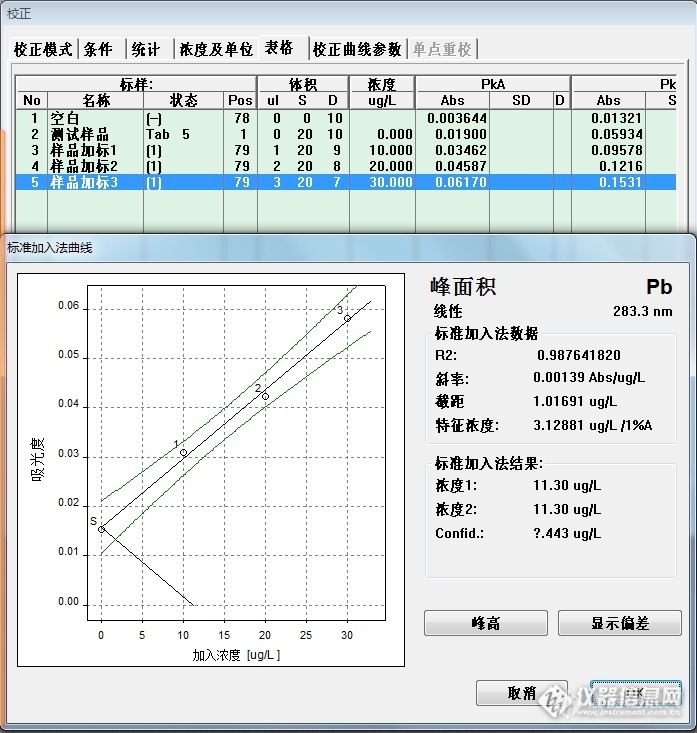
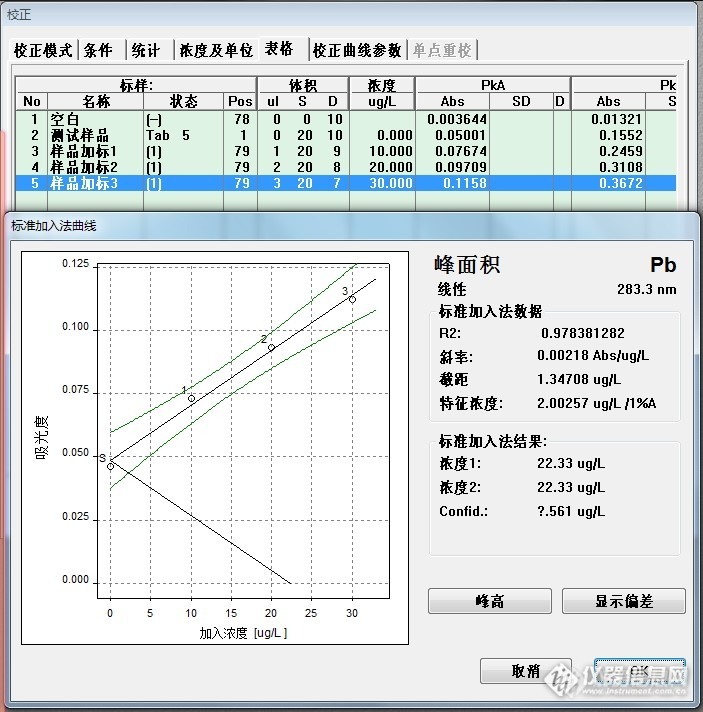
盲样测定,加标量分别为:10ug/L,20 ug/L,30 ug/L,数据如下图13:
![]()
图13
图13 样品说明:
No1-No5:样品消化空白及标系
No6-No10:盲样442及标系
No12-No15:盲样488及标系(经过预试验,盲样488稀释了4倍)
1%HNO
3空白Abs均值:峰面积Pa:0.0036435,峰高Ph:0.01321
线性回归如下:
![]()
图14
![]()
图15
![]()
图16
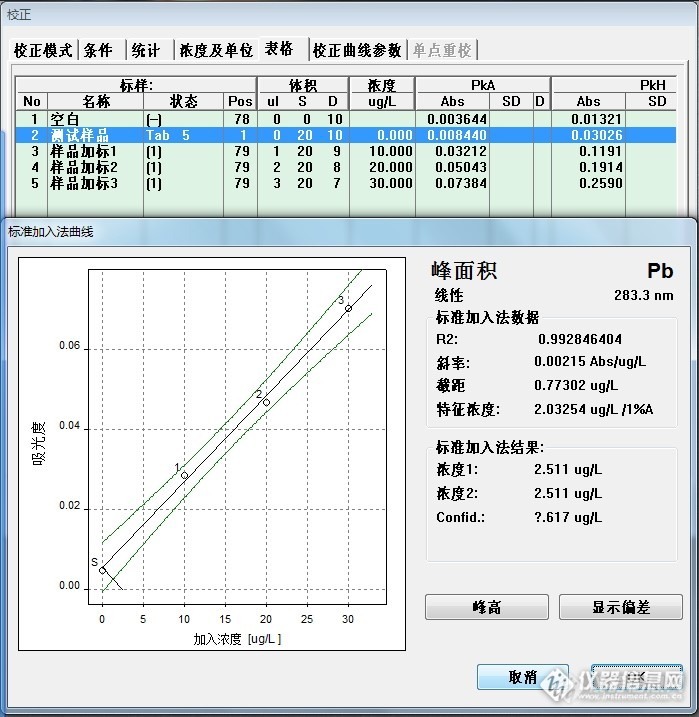
样品消化空白:2.511ug/L
442:11.30 ug/L
488:22.33 ug/L*4=89.32 ug/L
代入公式计算:X=(c-c
0)*v/(1000*m); v=25ml,m=1.00g
442:0.22 mg/Kg
488:2.17 mg/Kg
总结:
1,对于大葱样等含基体干扰的样品采用2% NH4H2PO4+1%Mg(NO3)2作为基改剂比单一的1% NH4H2PO4测定效果要好,但是对不含基体的标液而言影响不大。
2,对于不同的样品最好能针对具体样品做升温程序的优化,这样更具有针对性。所以当日常的程序无法测定时,不要急着把样品处理掉,并不一定就是消化的问题。
写完了,有不足或是不对的地方还请各位多多指教!